A comparative analysis of the functionalisation of unactivated cycloalkanes using alkynes and either sunlight or a photochemical reactor
Roisin A.
Doohan
and
Niall W. A.
Geraghty
*
Chemistry Department, National University of Ireland, Galway, Co. Galway, Ireland. E-mail: niall.geraghty@nuigalway.ie; Fax: +353-91-525700; Tel: +353-91-524411, Extn 2474
First published on 10th December 2004
Abstract
Using a standard mercury vapour lamp or sunlight, the synthetically difficult task of introducing functionality into unactivated cycloalkanes through C–C bond formation is accomplished in the presence of a soluble or supported photomediator and an alkyne bearing an electron-withdrawing group. The reaction involves the regiospecific addition of a photochemically generated cycloalkyl radical to the β-carbon of the alkyne. The use of solar radiation and a potentially recyclable polymer-bound photomediator for this fundamentally important synthetic process is particularly attractive from the clean/green chemistry perspective.
Introduction
Carbon–carbon bond formation is a fundamental synthetic process that generally involves a carbon atom which is activated by the presence of an adjacent functional group. The absence of functional groups on saturated hydrocarbons such as cycloalkanes, and ethers such as THF and dioxolanes, renders these compounds challenging with respect to their chemical transformation.1,2 The other problems encountered in activating these systems include achieving regioselectivity and preventing over-reaction of the initially functionalised alkane or ether. These functionalisations have been previously carried out using the reaction of electrophilic benzotriazole reagents with 1,3-dioxolane,3 the Gif oxidation procedure devised by Barton4 and reactions which involve either the direct or indirect generation of alkyl and ether radicals. The indirect generation of cycloalkyl radicals, and ultimately the functionalisation of cycloalkanes, has been achieved in numerous ways through the use of metal catalysts5 as well as appropriate radical precursors. These methods include the reaction of alkyl halides with tributyltin hydride6 or tris(trimethyl)silane,7 the oxyalkylation of cyclohexane and cyclooctane,8 the ethylation and vinylation of cyclohexane using polyoxotungstates as catalytic photoinitiators,9 the photolysis of alkylcobalt compounds,10 and the chain-transfer reaction of acetylenic triflones using AIBN.11 The functionalisation of unactivated systems by the direct generation of alkyl radicals has been achieved through the use of peroxides and a variety of other radical initiators. In this context the photochemical generation of radicals is particularly attractive as it avoids the use of metal catalysts and toxic reagents. In the case of cycloalkanes the peroxide initiated reactions include alkylation using di-tert-butyl peroxide,12 chlorination using a tert-butoxy radical,1 and oxidation using tert-butyl hydroperoxide.13 Most of these processes have been extended to include cyclic ethers, examples being the alkylation3 of 1,3-dioxolane, and the alkenylation, allylation and vinylation of THF by reaction with acetylenic triflones.14–17 The production of SO2 and the need to synthesise the reagents required, renders the reactions involving triflones, and those involving chloroethylsulfonyl oxime ethers18 which have been used to obtain acylated derivatives of cycloalkanes and ethers, problematic.The photochemical generation of radicals such as cyclopentyl, cyclohexyl and adamantyl has been achieved by both mercury19,20 and ketone photomediation. Functionalisations carried out in this way include acylation of cycloalkanes and sulfination of cycloalkanes to afford sulfinic acid derivatives,21 alkylation of ketene dithioacetal S,S-dioxides,22 acetylation of adamantane,23 the synthesis of β-cycloalkylnitriles from α,β-unsaturated nitriles,24 and the photochemical decomposition of ethyl azidoformate in cyclohexane producing cyclohexylurethane.25 Indeed the chain transfer reaction of acetylenic triflones with ethers and cycloalkanes has also been shown to proceed efficiently using irradiation in place of AIBN.11 It is the solar chemistry version of the photochemical production of alkyl radicals from alkanes using ketones, which is of relevance to the work described in this paper. This method of hydrogen abstraction occurs as a result of the absorption of a photon by a carbonyl group, usually a ketone, which subsequently exhibits alkoxy radical character in its n,π* triplet state. The excited ketone, for example benzophenone, is then able to abstract a hydrogen atom from a cycloalkane producing a cycloalkyl radical which can undergo a number of reactions including recombination, disproportionation or further hydrogen abstraction. We have shown previously that the cycloalkyl radical generated in this way can undergo Michael-type addition to an electron-deficient alkyne26 (Scheme 1), behavior which is consistent with its nucleophilic character.27 This paper focuses on the possibility of carrying out such a functionalisation under solar conditions and reports results which allow the solar photochemistry approach to be compared to the use of a conventional photochemical reactor.
 | ||
| Scheme 1 | ||
Reports of the photoreactions of alkynes with cycloalkanes first appeared in 1969 when Buchi and Feairheller,28 and independently Grovenstein et al.,29 described the irradiation of cyclohexane solutions of ethyl propiolate and dimethyl acetylenedicarboxylate (DMAD). These direct photoreactions gave products resulting from the insertion of ethyl propiolate and DMAD into the C–H bond of cyclohexane in yields of 5% and 10%, and reaction times of 24 h and 14 days, respectively. The cyclohexyl group was shown to add selectively to the β-carbon of ethyl propiolate. In 1993 Metzger and Blumenstein30 investigated the formation of Z and E alkenes in a thermally initiated radical chain reaction of alkynes with cycloalkanes and again found that the cyclohexyl group added regiospecifically to the unsubstituted end of the alkyne triple bond. As we have shown, the use of a photomediator has a dramatic effect on the C–C bond forming reaction between alkynes and cycloalkanes in terms of reaction times and yields. Initial studies found benzophenone to be one of the most efficient photomediators. In hexane it has an absorption band at 346 nm (log ε = 2.1), with a shoulder at 361 nm (log ε = 2.0),31 and thus can be excited electronically using sunlight. The ease with which cycloalkyl radicals can be produced in this way and the fact that they subsequently undergo C–C bond forming reactions with alkynes under these conditions, coupled with the possibility of carrying out the reaction using solar irradiation, makes the process synthetically important and environmentally attractive. The programme of work carried out at the Plataforma Solar de Almeria (PSA) which is described here, provides experimental data, which allow the use of a standard photochemical reactor and solar radiation to be compared. The possibility of using a supported, and thus potentially recyclable, photomediator was also considered.
Results and discussion
Experiments involving cyclopentane and cyclohexane, and both methyl propiolate (MP) and DMAD were carried out under solar irradiation and using a Rayonet photochemical reactor (350 nm). These reactions involved benzophenone as a soluble photomediator (Table 1) or the supported photomediators 5 and 6 (Table 2).
| Reactiona | Rayonet reactor (350 nm) | Solar irradiation | ||||
|---|---|---|---|---|---|---|
| Time/hb |
Z![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) ∶E Ratioc ∶E Ratioc |
Yield (%)c | Time/hb |
Z∶E Ratioc |
Yield (%)c | |
| a Reaction conditions: benzophenone (0.43 mmol, 0.011 M), cycloalkane (40 ml), alkyne (3 mmol), dodecane as internal standard. b Time for complete consumption of the alkyne. c GC. | ||||||
| C5H10 | 2.5 | 1∶1.7 |
95 | 3.7 | 1∶1.8 |
77 |
HC![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C–CO2CH3 C–CO2CH3 |
||||||
| C6H12 | 4.5 | 1∶1.5 |
59 | 6.5 | 1∶1.8 |
48 |
| HCC–CO2CH3 |
||||||
| C5H10 | 2.5 | 12∶1 |
74 | 4.0 | 28∶1 |
78 |
| H3CO2C–CC–CO2CH3 |
||||||
| C6H12 | 3.75 | 13∶1 |
64 | 4.5 | 10.5∶1 |
55 |
| H3CO2C–CC–CO2CH3 |
||||||
| Reaction | Rayonet reactor (350 nm) | Solar irradiation | ||||
|---|---|---|---|---|---|---|
| Time/ha |
Z∶E Ratiob |
Yield (conversion) (%)b | Time/ha |
Z∶E Ratiob |
Yield (conversion) (%)b | |
| a Total reaction time. b GC. c Reaction conditions: 5 (2 g), cycloalkane (40 ml), alkyne (3 mmol), dodecane as internal standard. d Reaction conditions: as before, 6 (2 g). e Reaction conditions: as before, 5 (2 × 1 g). | ||||||
| C5H10 | 7.5 | 1∶1.7 |
57(100)c | 55 | 1∶1.6 |
20(71)e |
| HCC–CO2CH3 |
18 | 1∶1.5 |
62(100)d | 12 | 1∶1.7 |
29(61)d |
| C6H12 | 40 | 1∶1.4 |
31(72)c | 13.5 | 1∶1.6 |
3(24)c |
| HCC–CO2CH3 |
||||||
| C5H10 | 55 | 2.5∶1 |
42(100)c | 52 | 2.5∶1 |
34(83)e |
| H3CO2C–CC–CO2CH3 |
||||||
| C6H12 | 15 | 2.2∶1 |
49(100)c | 6.5 | 1.5∶1 |
7(4)c |
| H3CO2C–CC–CO2CH3 |
||||||
The reactions of cyclopentane and cyclohexane with MP in the presence of benzophenone, resulted in the formation of mixtures of the (Z)- and (E)-propenoates, 1a and 1b
(Fig. 1 and Fig. 2), and 2a and 2b, respectively, with the (E)-isomer predominating (Scheme 1). No evidence of secondary photochemical isomerisation was observed, and thus it can be concluded that the Z∶E ratio reflects the stereoselectivity of the addition of the cycloalkyl radical to the alkyne. The only other low molecular weight products observed were the corresponding cycloalkanones and cycloalkanols, which are formed in trace amounts (GC-MS). The benzophenone mediated solar and Rayonet reactions of MP are initially very rapid but slow considerably towards the end. A further trend in these reactions is that the reactivity of cyclopentane is greater than that of cyclohexane.32,33

 | ||
| Fig. 1 Rayonet reaction of MP, cyclopentane and benzophenone. | ||
 | ||
| Fig. 2 Solar reaction of MP, cyclopentane and benzophenone. | ||
The reactions of cyclopentane and cyclohexane with the disubstituted alkyne, DMAD, resulted in the formation of the (Z)- and (E)-3-cycloalkyl-2-pentenedioates, 3a and 3b (Fig. 3 and Fig. 4), and 4a and 4b, respectively. However, in contrast to the MP reactions, both the solar and Rayonet reactions are relatively stereoselective as a result of a secondary cis/trans photoisomerisation process. DMAD was found to be slightly less reactive than MP, and the greater reactivity of the cyclopentane ring system was again apparent.
 | ||
| Fig. 3 Rayonet reaction of DMAD, cyclopentane and benzophenone. | ||
 | ||
| Fig. 4 Solar reaction of DMAD, cyclopentane and benzophenone. | ||
Further analysis of the data obtained (Table 1), indicates that although the solar reactions using a soluble photomediator require longer reaction times than those carried out in the photochemical reactor, they give comparable GC yields in most cases, and indeed in one case, the reaction of cyclopentane and DMAD, the yield obtained is slightly higher. Surprisingly, the Z∶E ratio of the products obtained from cyclohexane and DMAD is lower when solar radiation is used, despite the longer reaction time. The reaction of cyclopentane and MP under solar irradiation conditions (Fig. 2) is initially faster than the corresponding reaction in a Rayonet reactor (Fig. 1) whereas the opposite occurs in the reaction of DMAD and cyclopentane.
The use of the supported photomediators 5 and 6 leads to significantly increased reaction times, and in general the supported photomediators perform poorly in comparison to the soluble benzophenone, particularly under solar irradiation (Table 2). A uniform suspension of the supported mediator was achieved using a magnetic stirrer and the settling times of the solid when stirring was stopped suggested that mechanical breakdown of the support was not a problem. The formation of the alkenes using MP in the Rayonet reactor and using solar radiation leads, as before, to the predominant formation of the (E)-isomers, 1b and 2b. The (Z)-isomers, 3a and 4a are again the major products formed when DMAD is used. However in the latter case, despite the long reaction times, the Z∶E ratio is much smaller than that observed for the corresponding reactions involving soluble benzophenone. The time course data for the solar reactions of cyclopentane with MP (Fig. 5) and DMAD (Fig. 6) show that the supported photomediator 5 becomes rapidly deactivated. On addition of a further portion of the resin bound mediator the reaction recommences, but soon ceases again. The concept that the photomediator is becoming inactive is supported by the fact that the photosensitized isomerisation of the primary photoinsertion products, normally observed with DMAD, does not occur, despite the long reaction times, as evidenced by the Z∶E ratios (Table 2).
 | ||
| Fig. 5 Solar reaction of MP and cyclopentane in the presence of 5. | ||
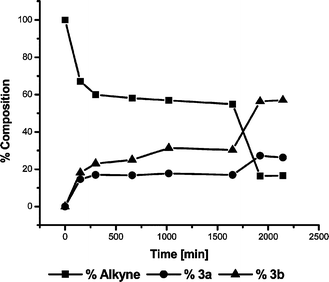 | ||
| Fig. 6 Solar reaction of DMAD and cyclopentane in the presence of 5. | ||
One possible reason for this deactivation is the formation of a polymeric coating on the surface of the particles of supported photomediator. Alternatively, deactivation may occur as a result of photochemical reactions involving the polystyrene resin. A significant loss of reactivity is also observed in solar reactions involving cyclohexane and the Merrifield resin bound benzophenone 5 (Fig. 7), which with MP resulted in 24% conversion, and a yield of only 3%, after 13.5 h.
 | ||
| Fig. 7 Solar reaction of MP, cyclohexane and 5. | ||
The aminopropylsilica bound benzophenone 6 is a more effective photomediator for these reactions. The solar irradiation of a cyclopentane solution of MP containing 6 (Fig. 8) gives a yield of 29% (61% conversion of the alkyne) after 12 h and 62% (100% conversion) after 18 h irradiation in a Rayonet reactor. The supported benzophenone 6 appears to be more robust as although the reactions are slow they do not stop altogether. A 3 to 7 fold increase in the amount of cycloalkanols and cycloalkanones occurs when supported photomediators are used under solar conditions or in the Rayonet reactor. These products are formed by reaction of cycloalkyl radicals with molecular oxygen and their formation would be expected if other reactions of the cycloalkyl radical, such as those with the alkyne, were inefficient. This suggests that accessibility of the alkyne to the cycloalkyl radical, rather than problems in generating the radical, may be responsible for the poor performance of the supported photomediators. In the wider context of the overall mechanism, the GC monitoring (using dodecane as internal standard) of the reactions involving benzophenone clearly shows that its concentration does not change during the course of the reaction. It would thus appear that the reaction is based on an inefficient chain process, involving the alkyne and cycloalkyl radicals, which is terminated relatively quickly by hydrogen transfer from a benzhydroxyl radical.
 | ||
| Fig. 8 Solar reaction of MP, cyclopentane and 6. | ||
Conclusions
The fundamentally important process of functionalising saturated cycloalkanes through C–C bond formation has been achieved through the use of a photomediator and solar radiation. This method has the additional environmental advantage that it does not involve the use of metals such as tin,6 or the production of by-products such as SO2,11,18 which are features of other methods. The reaction is regiospecific with cycloalkane insertion occurring at the unsubstituted end of the alkyne. Although the reaction of monosubstituted alkynes is only moderately stereoselective, the involvement of a photomediator controlled secondary photoisomerisation leads to high Z∶E ratios with disubstituted alkynes such as DMAD. In keeping with the results obtained with other systems, cyclopentane was found to be more reactive than cyclohexane. The use of solar radiation with benzophenone, a soluble photomediator, gave results that were broadly comparable to those obtained with a conventional photochemical reactor. A Merrifield resin supported form of benzophenone, 5, was less efficient under both standard UV and solar irradiation, the reduction being most marked in the latter case in which the photomediator appeared to become rapidly deactivated. More promising results were obtained with the supported photomediator 6, a 3-aminopropyl functionalised silica gel. Overall the C–H activation process described in this paper, involving solar radiation and a potentially recyclable photomediator, is very attractive in the context of clean/green chemistry.
Experimental
The cycloalkanes and alkynes were distilled before use. The photochemical reactions were carried out in cylindrical Pyrex tubes. A Rayonet Photochemical Reactor, RPR-100, encompassing sixteen 350 nm mercury lamps was used. IR spectra were measured on a Perkin Elmer Spectrum 1 FT-IR. 1H NMR and 13C NMR were measured on a Jeol JNM-LA400 spectrometer at probe temperatures using CDCl3 as solvent and TMS as the internal standard. All GC analyses at NUI, Galway, were carried out on an RTX-5 (Restek) column, while at PSA a HP-5 (Agilent) column was used. GC-MS analyses were carried out on a Micromass GCT spectrometer together with an Agilent 6890 capillary gas chromatograph equipped with a HP-5 column.General procedure for photochemical reactions using benzophenone as photomediator
A solution of benzophenone (0.08 g, 0.43 mmol) and dodecane (0.37 g, 2 mmol) in cycloalkane (40 ml) was degassed with N2 for 20 min in a cylindrical Pyrex tube. The alkyne (3 mmol) was added and the stirred solution was irradiated using 350 nm lamps until all the alkyne had been consumed (GC). The crude product mixture contained the cis and trans alkene products, and also trace amounts of the corresponding cycloalkanols and cycloalkanones (GC-MS). Subsequent to the removal of excess solvent the mixture was adsorbed onto silica (50 g) and eluted with bp 40–60 °C petroleum ether–ether (95∶5). All products were obtained as clear, sweet smelling oils. The solar reactions carried out at PSA involved placing the cylindrical Pyrex tubes on a flat sheet of aluminium foil which was angled towards the sun. Exposure to solar radiation continued until all of the alkyne had been consumed (GC). A series of experiments involving different cycloalkanes and alkynes were carried out in a Rayonet reactor and under solar irradiation using the same general reaction conditions (Table 1).
General procedure for photochemical reactions using a supported photomediator
The cycloalkane (40 ml) and supported photomediator were degassed for 20 min using N2. Dodecane (0.37 g, 2 mmol) and the alkyne (3 mmol) were added and the mixture was stirred vigorously and irradiated using 350 nm lamps. GC analysis of the crude product mixture indicated the presence of the expected products. The supported photomediator was washed with dichloromethane (100 ml), ether (100 ml), ethanol (100 ml) and methanol (100 ml). Removal of the solvent gave the products together with trace amounts of the cycloalkanols and cycloalkanones. The support was then dried and weighed resulting in 99% recovery.The corresponding solar reactions at PSA were carried out as indicated above. In some cases further quantities of the supported photomediator were added and its effect monitored. A series of experiments involving different cycloalkanes and alkynes were carried out in a Rayonet reactor and under solar irradiation using the same general reaction conditions (Table 2).
Synthesis of supported photomediators
Analytical data
1a, Methyl (Z)-3-cyclopentyl-2-propenoate.26 IR (cm−1): 1721 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O), 1644 (CC), 1176 and 1151 (C–O), 819 (CHCH). 1H NMR: δ 1.26 (ms, 2H), 1.61–1.69 (ms, 4H), 1.90 (ms, 2H), 3.67 (m, 1H, CH–CHCH), 3.70 (s, 3H, OCH3), 5.67 (d, 1H, CH–CHCH, Jcis
= 11.2 Hz), 6.12 (t, 1H, CH–CHCH, Jvic
=
Jcis
= 11.2 Hz). 13C NMR: δ 166.9, 155.6, 117.4, 50.8, 39.0, 33.3, 25.5. m/z
(%) 154 (18), 122 (20), 111 (11), 95 (18), 94 (100), 87 (30), 79 (48), 67 (51).
O), 1644 (CC), 1176 and 1151 (C–O), 819 (CHCH). 1H NMR: δ 1.26 (ms, 2H), 1.61–1.69 (ms, 4H), 1.90 (ms, 2H), 3.67 (m, 1H, CH–CHCH), 3.70 (s, 3H, OCH3), 5.67 (d, 1H, CH–CHCH, Jcis
= 11.2 Hz), 6.12 (t, 1H, CH–CHCH, Jvic
=
Jcis
= 11.2 Hz). 13C NMR: δ 166.9, 155.6, 117.4, 50.8, 39.0, 33.3, 25.5. m/z
(%) 154 (18), 122 (20), 111 (11), 95 (18), 94 (100), 87 (30), 79 (48), 67 (51).
1b, Methyl (E)-3-cyclopentyl-2-propenoate.26,36 IR (cm−1): 1721 (CO), 1654 (CC), 1171 and 1148 (C–O), 983 (CHCH). 1H NMR: δ 1.36 (ms, 2H), 1.60–1.69 (ms, 4H), 1.82 (ms, 2H), 2.58 (m, 1H, CH–CHCH), 3.70 (s, 3H, OCH3), 5.81 (d, 1H, CH–CHCH, Jtrans
= 15.6 Hz), 6.96 (dd, 1H, CH–CHCH, Jvic
= 7.8 Hz and Jtrans
= 15.6 Hz). 13C NMR: δ 167.2, 153.6, 118.7, 51.1, 42.6, 32.2, 25.0. m/z
(%) 154 (100), 122 (31), 111 (27), 95 (28), 94 (75), 87 (27), 79 (29), 67 (14).
2a, Methyl (Z)-3-cyclohexyl-2-propenoate.30 IR (cm−1): 1725 (CO), 1652 (CC), 1181 and 1175 (C–O), 820 (CHCH). 1H NMR: δ 0.99–1.30 (ms, 5H), 1.54–1.72 (ms, 5H), 3.24 (m, 1H, CH–CHCH), 3.63 (s, 3H, OCH3), 5.58 (d, 1H, CH–CHCH, Jcis
= 11.3 Hz), 5.96 (t, 1H, CH–CHCH, Jvic
=
Jcis
= 11.3 Hz). 13C NMR: δ 166.7, 155.9, 117.1, 50.9, 37.3, 32.2, 25.8, 25.4.
2b, Methyl (E)-3-cyclohexyl-2-propenoate.30 IR (cm−1): 1721 (CO), 1650 (CC), 1195 and 1169 (C–O), 983 (CHCH). 1H NMR: δ 1.12–1.34 (ms, 5H), 1.65–1.77 (ms, 5H), 2.13 (m, 1H, CH–CHCH), 3.72 (s, 3H, OCH3), 5.78 (d, 1H, CH–CHCH, Jtrans
= 15.7 Hz), 6.91 (dd, 1H, CH–CHCH, Jvic
= 6.9 Hz and Jtrans
= 15.7 Hz). 13C NMR: δ 167.4, 154.4, 118.3, 51.2, 40.3, 32.2, 25.7, 25.5.
3a, Dimethyl (Z)-2-cyclopentyl-2-butenedioate.26 IR (cm−1): 1722 (CO), 1642 (CC), 1163 (C–O). 1H NMR: δ 1.50–1.71 (ms, 6H), 1.71–1.87 (ms, 2H), 2.76 (m, 1H, CH–CHCH), 3.71 (s, 3H, OCH3), 3.83 (s, 3H, OCH3), 5.80 (s, 1H, CCH). 13C NMR: δ 169.0, 165.1, 154.6, 116.4, 51.8, 51.4, 44.0, 30.5, 24.4.
4a, Dimethyl (Z)-2-cyclohexyl-2-butenedioate.26 IR (cm−1): 1723 (CO), 1644 (CC), 1165 (C–O). 1H NMR: δ 1.14–1.31 (ms, 5H), 1.67–1.90 (ms, 5H), 2.30 (m, 1H, CH–CHCH), 3.71 (s, 3H, OCH3), 3.83 (s, 3H, OCH3), 5.77 (s, 1H, CCH). 13C NMR: δ 169.3, 165.6, 156.3, 116.1, 52.0, 51.6, 42.2, 30.8, 25.8, 25.5.
Acknowledgements
The funds provided to R.A.D by Enterprise Ireland, Donegal County Council and the IHP programme of EU-DGXII are gratefully acknowledged. Also gratefully acknowledged is the support received from the staff of Plataforma Solar de Almeria, Spain.References
- P. A. MacFaul, I. W. C. E. Arends, K. U. Ingold and D. D. M. Wagner, J. Chem. Soc., Perkin Trans. 2, 1997, 135 RSC.
- A. F. Shilov and G. B. Shul'pin, Chem. Rev., 1997, 97, 2879 CrossRef CAS.
- A. R. Katritzky, H. H. Odens and M. V. Voronkov, J. Org. Chem., 2000, 65, 1886 CrossRef CAS.
- D. H. R. Barton, M. J. Gastiger and W. B. Motherwell, J. Chem. Soc., Chem. Commun., 1983, 41 RSC.
- G. Dyker, Angew. Chem., Int. Ed., 1999, 38, 1699 CrossRef CAS.
- D. P. Curran, Synthesis, 1988, 417 CrossRef CAS.
- P. Dowd and W. Zhang, Chem. Rev., 1993, 93, 2091 CrossRef CAS.
- T. Hara, T. Iwahama, S. Sakaguchi and Y. Ishii, J. Org. Chem., 2001, 66, 6425 CrossRef CAS.
- B. S. Jaynes and C. L. Hill, J. Am. Chem. Soc., 1993, 115, 12212 CrossRef CAS.
- G. Pattenden, Chem. Soc. Rev., 1988, 17, 361 RSC.
- J. Gong and P. L. Fuchs, J. Am. Chem. Soc., 1996, 118, 4486 CrossRef.
- I. Tabushi and K. Fukunishi, J. Org. Chem., 1974, 39, 3748 CrossRef CAS.
- F. Launay, A. Roucoux and H. Patin, Tetrahedron Lett., 1998, 39, 1353 CrossRef CAS.
- J. Xiang and P. L. Fuchs, J. Am. Chem. Soc., 1996, 11986 CrossRef CAS.
- J. Xiang, W. Jiang, J. Gong and P. L. Fuchs, J. Am. Chem. Soc., 1997, 119, 4123 CrossRef CAS.
- J. Xiang, W. Jiang and P. L. Fuchs, Tetrahedron Lett., 1997, 38, 6635 CrossRef CAS.
- J. Xiang, J. Evarts, A. Rivkin, D. P. Curran and P. L. Fuchs, Tetrahedron Lett., 1998, 39, 4163 CrossRef CAS.
- S. Kim, N. Kim, W. Chung and C. H. Cho, Synlett, 2001, 937 CrossRef CAS.
- S. H. Brown and R. H. Crabtree, Tetrahedron Lett., 1987, 28, 5599 CrossRef CAS.
- S. H. Brown and R. H. Crabtree, J. Am. Chem. Soc., 1989, 111, 2935 CrossRef CAS.
- R. R. Ferguson and R. H. Crabtree, J. Org. Chem., 1991, 56, 5503 CrossRef CAS.
- A. M. Gonzalez-Cameno, M. Mella, M. Fagnoni and A. Albini, J. Org. Chem., 2000, 65, 297 CrossRef CAS.
- I. Tabushi, S. Kojo and K. Fukunishi, J. Org. Chem., 1978, 43, 2370 CrossRef CAS.
- A. M. Cardarelli, M. Fagnoni, M. Mella and A. Albini, J. Org. Chem., 2001, 66, 7320 CrossRef CAS.
- W. Lwowski and T. W. Mattingly, J. Am. Chem. Soc., 1965, 87, 1947 CrossRef CAS.
- N. W. A. Geraghty and J. J. Hannan, Tetrahedron Lett., 2001, 42, 3211 CrossRef CAS.
- B. Giese and S. Lachhein, Angew. Chem., Int. Ed. Engl., 1982, 21, 768 CrossRef.
- G. Buchi and S. H. Feairheller, J. Org. Chem., 1969, 34, 609 CrossRef.
- E. Grovenstein, T. C. Campbell and T. Shibata, J. Org. Chem., 1969, 34, 2418 CrossRef.
- J. O. Metzger and M. Blumenstein, Chem. Ber., 1993, 126, 2493 CrossRef CAS.
- CRC Handbook of Chemistry and Physics, ed. D. R. Lide, CRC Press, Boca Raton, FL, 78th edn., 1997 Search PubMed.
- J. G. Traynham and Y. S. Lee, J. Am. Chem. Soc., 1974, 96, 3590 CrossRef CAS.
- E. L. Eliel and S. H. Wilen, Stereochemistry of Organic Compounds, John Wiley & Sons, New York, 1994, p.769 Search PubMed.
- E. C. Blossey and D. C. Neckers, Tetrahedron Lett., 1974, 15, 323 CrossRef.
- M. Ayadim and J. P. Soumillion, Tetrahedron Lett., 1996, 37, 381 CrossRef CAS.
- R. D. Hubbard and B. L. Miller, J. Org. Chem., 1998, 63, 4143 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2005 |