Global partitioning of NOx sources using satellite observations: Relative roles of fossil fuel combustion, biomass burning and soil emissions
Lyatt
Jaeglé
a,
Linda
Steinberger
a,
Randall V.
Martin
bc and
Kelly
Chance
c
aDepartment of Atmospheric Sciences, University of Washington, Seattle, Washington, USA. E-mail: jaegle@atmos.washington.edu
bDepartment of Physics and Atmospheric Science, Dalhousie University, Halifax, Canada
cHarvard-Smithsonian Center for Astrophysics, Cambridge, Massachusetts, USA
First published on 11th May 2005
Abstract
We use space-based observations of NO2 columns from the Global Ozone Monitoring Experiment (GOME) to derive monthly top-down NOx emissions for 2000 via inverse modeling with the GEOS-CHEM chemical transport model. Top-down NOx sources are partitioned among fuel combustion (fossil fuel and biofuel), biomass burning and soils by exploiting the spatio-temporal distribution of remotely sensed fires and a priori information on the location of regions dominated by fuel combustion. The top-down inventory is combined with an a priori inventory to obtain an optimized a posteriori estimate of the relative roles of NOx sources. The resulting a posteriori fuel combustion inventory (25.6 TgN year−1) agrees closely with the a priori (25.4 TgN year−1), and errors are reduced by a factor of 2, from ±80% to ±40%. Regionally, the largest differences are found over Japan and South Africa, where a posteriori estimates are 25% larger than a priori. A posteriori fuel combustion emissions are aseasonal, with the exception of East Asia and Europe where winter emissions are 30–40% larger relative to summer emissions, consistent with increased energy use during winter for heating. Global a posteriori biomass burning emissions in 2000 resulted in 5.8 TgN (compared to 5.9 TgN year−1 in the a priori), with Africa accounting for half of this total. A posteriori biomass burning emissions over Southeast Asia/India are decreased by 46% relative to a priori; but over North equatorial Africa they are increased by 50%. A posteriori estimates of soil emissions (8.9 TgN year−1) are 68% larger than a priori (5.3 TgN year−1). The a posteriori inventory displays the largest soil emissions over tropical savanna/woodland ecosystems (Africa), as well as over agricultural regions in the western U.S. (Great Plains), southern Europe (Spain, Greece, Turkey), and Asia (North China Plain and North India), consistent with field measurements. Emissions over these regions are highest during summer at mid-latitudes and during the rainy season in the Tropics. We estimate that 2.5–4.5 TgN year−1 are emitted from N-fertilized soils, at the upper end of previous estimates. Soil and biomass burning emissions account for 22% and 14% of global surface NOx emissions, respectively. We infer a significant role for soil NOx emissions at northern mid-latitudes during summer, where they account for nearly half that of the fuel combustion source, a doubling relative to the a priori. The contribution of soil emissions to background ozone is thus likely to be underestimated by the current generation of chemical transport models.
1. Introduction
Human activities have led to a three- to six-fold increase in nitrogen oxides (NOx = NO + NO2) emissions since pre-industrial times.1 These large anthropogenic emissions, mostly resulting from fossil fuel combustion and biomass burning, are superimposed on natural sources of NOx, which include microbial processes in soils, lightning and transport from the stratosphere.2 While the contribution of fossil fuel combustion to the global NOx budget is relatively well known (20–24 TgN year−1), estimates of the other surface NOx sources remain highly uncertain: biomass burning (3–13 TgN year−1) and soil-atmosphere exchange (4–21 TgN year−1).3Improved estimates of NOx emissions are crucial to better understanding regional and global ozone air pollution, acid deposition, and climate change. Current NOx inventories are based on “bottom-up” estimates which aggregate information from diverse sources: country-by-country statistics on fuel and land use, agricultural data, estimates of burned areas, observations of NOx emissions ratios in pollution plumes, and local measurements of soil fluxes.
Space-based observations of NO2 columns from the Global Ozone Monitoring Experiment (GOME)4,5 on board the European Remote Sensing (ERS-2) satellite can provide independent “top-down” constraints on global surface NOx emissions.6 Martin et al.7 demonstrated how GOME tropospheric NO2 columns can be related to surface NOx emissions via inverse modeling, thus deriving a top-down emission inventory. When combined with a bottom-up inventory, these top-down emissions can reduce significantly the uncertainties in NOx emissions.
In this study, we build on the work of Martin et al.,7 and demonstrate how added information from satellite observations of fires and from the location of fossil fuel dominated regions can help to partition GOME top-down NOx sources among fuel combustion (fossil fuel and biofuel), biomass burning, and soil emissions. This allows us to examine the magnitude, geographical distribution and seasonal variations of these individual sources and compare them to bottom-up estimates. We previously applied this method to examine NOx sources over Africa.8 Here, we improve on this method and apply it to the global NOx budget for the year 2000.
In section 2 we describe the bottom-up NOx emissions inventory used as the a priori in our analysis. In section 3, we infer top-down surface NOx emissions from GOME NO2 column observations, and describe a means for partitioning these emissions. The partitioned top-down and a priori inventories are combined into an optimized a posteriori inventory in section 4. We discuss our results in section 5, and compare our global a posteriori partitioning of surface NOx sources to previous studies in section 6. Conclusions are given in section 7.
2. A priori NOx emission inventory
We use NOx emissions from the GEOS-CHEM global model of tropospheric chemistry9 as our a priori global NOx inventory. This inventory was previously described7 and includes emissions from fossil fuel (23.3 TgN year−1), biofuel10 (2.2 TgN year−1), biomass burning11,12 (5.9 TgN year−1), soils (5.3 TgN year−1), aircraft (0.05 TgN year−1), stratosphere-troposphere exchange (0.1 TgN year−1), and lightning (3.5 TgN year−1). More specifically, anthropogenic emissions are from the Global Emission Inventory Activity (GEIA),13 scaled to 1998 as described in Bey et al.9 The biomass burning inventory includes interannual and seasonal variability as derived by Duncan et al.,12 using satellite observations of fires from the Along Track Scanning Radiometer (ATSR) and aerosols from the Total Ozone Monitoring Spectrometer (TOMS). The algorithm of Yienger and Levy14 is applied to calculate soil emissions, with the canopy reduction factor described in Wang et al.15 All emissions are on a 2° latitude by 2.5° longitude horizontal resolution. Tables 1–4 summarize the surface emission inventory for the individual regions shown in Fig. 1. In addition, the middle column of Fig. 2 illustrates the spatial distribution of these surface sources. Here and in the rest of the paper we combine fossil fuel and biofuel emissions into a single “fuel combustion” category. | ||
| Fig. 1 Regions used in Tables 1–4 and Figs. 5 and 6. | ||
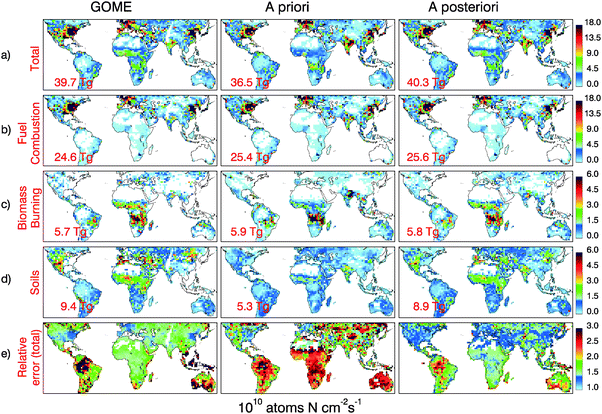 | ||
| Fig. 2 Annual surface NOx emissions for 2000: GOME top-down (left panels), a priori (middle panels) and a posteriori (right panels) inventories. Total surface NOx emissions, shown on the top panels (a), are partitioned among: (b) fuel combustion, (c) biomass burning, and (d) soil emissions. All emissions are in 1010 atoms N cm−2 s−1 and the global annual emissions are indicated at the bottom left of each panel in TgN yr−1. The bottom panels (e) display the relative errors (unitless) for total surface NOx emissions. | ||
| Total surface NOx emissions/TgN year−1 | ||||
|---|---|---|---|---|
| A priori | GOME | A posteriori | M2003b | |
| a Surface NOx emissions include fossil fuel combustion, biofuel combustion, biomass burning and soil emissions. Relative errors are indicated in parentheses. b A posteriori emissions inventory from Martin et al.7 based on GOME 1996–1997 NO2 columns. | ||||
| (a) United States | 6.8 (1.4) | 8.3 (1.5) | 7.4 (1.3) | 7.3 |
| (b) Europe | 5.5 (1.7) | 6.2 (1.5) | 6.2 (1.4) | 6.3 |
| (c) East Asia | 5.3 (2.0) | 6.6 (1.5) | 6.3 (1.5) | 6.0 |
| (d) Japan | 0.54 (1.5) | 0.81 (1.5) | 0.67 (1.3) | 0.73 |
| (e) Mideast | 1.1 (2.1) | 1.6 (1.5) | 1.3 (1.4) | 1.2 |
| (f) S. Africa | 0.36 (3.5) | 0.56 (1.6) | 0.48 (1.5) | 0.41 |
| (g) SE Asia/India | 4.9 (1.8) | 2.4 (2.0) | 4.3 (1.6) | 3.9 |
| (h) N. equatorial Africa | 2.2 (2.3) | 3.7 (1.6) | 3.3 (1.5) | 2.7 |
| (i) S. equatorial Africa | 2.4 (2.6) | 2.6 (1.7) | 2.5 (1.6) | 2.3 |
| (j) Central America | 1.7 (2.0) | 1.3 (1.9) | 1.5 (1.6) | 1.4 |
| (k) S. America | 2.7 (2.1) | 2.3 (1.9) | 2.8 (1.7) | 2.4 |
| (l) Australia | 1.0 (2.3) | 0.94 (2.2) | 1.2 (1.8) | 0.94 |
| Global | 36.5 (2.0) | 39.7 (1.6) | 40.3 (1.5) | 37.8 |
| Fuel combustiona emissions/TgN year−1 | ||||
|---|---|---|---|---|
| A priori | GOME | A posteriori | EDGAR 3.216 | |
| a Includes fossil fuel and biofuel emissions. Relative errors are indicated in parentheses. | ||||
| (a) United States | 6.3 (1.4) | 7.1 (1.5) | 6.4 (1.2) | 6.0 |
| (b) Europe | 4.9 (1.7) | 4.6 (1.6) | 4.9 (1.4) | 6.7 |
| (c) East Asia | 4.8 (2.0) | 5.3 (1.7) | 5.2 (1.5) | 4.7 |
| (d) Japan | 0.53 (1.6) | 0.80 (1.5) | 0.66 (1.3) | 0.81 |
| (e) Mideast | 0.93 (2.1) | 1.1 (1.6) | 0.93 (1.4) | 0.62 |
| (f) S. Africa | 0.27 (3.9) | 0.39 (1.5) | 0.33 (1.4) | 0.38 |
| (g) SE Asia/India | 3.2 (1.6) | 1.8 (2.0) | 3.0 (1.4) | 3.2 |
| (h) N. equatorial Africa | 0.40 (1.5) | 0.39 (1.6) | 0.39 (1.3) | 0.44 |
| (i) S. equatorial Africa | 0.18 (1.8) | 0.16 (1.9) | 0.18 (1.5) | 0.20 |
| (j) Central America | 1.0 (1.8) | 0.78 (1.7) | 0.89 (1.3) | 0.85 |
| (k) S. America | 1.0 (1.5) | 0.72 (1.8) | 1.0 (1.4) | 0.81 |
| (l) Australia | 0.31 (1.8) | 0.19 (2.2) | 0.31 (1.6) | 0.38 |
| Global | 25.4 (1.8) | 24.6 (1.6) | 25.6 (1.4) | 26.1 |
| Biomass burning emissions/TgN year−1 | ||||
|---|---|---|---|---|
| A priori | GOME | A posteriori | GWEM 1.441 | |
| (a) United States | 0.11 (3) | 0.17 (1.5) | 0.12 (1.7) | 0.05 |
| (b) Europe | 0.09 (3) | 0.30 (1.5) | 0.19 (1.7) | 0.10 |
| (c) East Asia | 0.13 (3) | 0.20 (1.5) | 0.18 (1.7) | 0.28 |
| (d) Japan | 0.00 (3) | 0.00 (1.5) | 0.00 (1.8) | 0.01 |
| (e) Mideast | 0.00 (3) | 0.03 (1.5) | 0.02 (1.5) | 0.00 |
| (f) S. Africa | 0.03 (3) | 0.07 (1.5) | 0.05 (1.8) | 0.10 |
| (g) SE Asia/India | 1.1 (3) | 0.28 (1.7) | 0.61 (2.5) | 0.16 |
| (h) N. equatorial Africa | 0.77 (3) | 1.3 (1.5) | 1.2 (1.5) | 1.4 |
| (i) S. equatorial Africa | 1.8 (3) | 1.8 (1.6) | 1.7 (1.5) | 1.4 |
| (j) Central America | 0.37 (3) | 0.25 (1.8) | 0.30 (2.0) | 0.04 |
| (k) S. America | 0.87 (3) | 0.81 (1.7) | 0.81 (1.9) | 0.16 |
| (l) Australia | 0.33 (3) | 0.38 (1.8) | 0.40 (1.9) | 0.43 |
| Global | 5.9 (3) | 5.7 (1.6) | 5.8 (1.8) | 5.0 |
| Soil emissions/TgN year−1 | |||
|---|---|---|---|
| A priori | GOME | A posteriori | |
| (a) United States | 0.41 (3) | 1.0 (1.5) | 0.86 (1.7) |
| (b) Europe | 0.48 (3) | 1.3 (1.5) | 1.1 (1.7) |
| (c) East Asia | 0.42 (3) | 1.1 (1.6) | 0.94 (1.7) |
| (d) Japan | 0.01 (3) | 0.0 (1.6) | 0.01 (2.6) |
| (e) Mideast | 0.17 (3) | 0.53 (1.6) | 0.40 (1.6) |
| (f) S. Africa | 0.06 (3) | 0.10 (2.0) | 0.10 (2.0) |
| (g) SE Asia/India | 0.59 (3) | 0.32 (2.2) | 0.65 (2.8) |
| (h) N. equatorial Africa | 0.98 (3) | 2.0 (1.7) | 1.7 (1.6) |
| (i) S. equatorial Africa | 0.41 (3) | 0.65 (1.9) | 0.64 (1.9) |
| (j) Central America | 0.34 (3) | 0.28 (2.0) | 0.35 (2.2) |
| (k) S. America | 0.77 (3) | 0.81 (2.1) | 1.0 (2.3) |
| (l) Australia | 0.37 (3) | 0.37 (2.5) | 0.47 (2.3) |
| Global | 5.3 (3) | 9.4 (1.7) | 8.9 (1.9) |
Following Martin et al.,7 we estimate the relative errors for the fuel combustion emissions as the ratio of a priori to EDGAR v3.2 1995 emissions.16 For biomass burning and soils we use an estimate of the relative error of a factor of 3, reflecting the range of global estimates for both sources.1 The overall a priori error is smaller than 1.5 (±50%) over regions dominated by fuel combustion emissions but increases to values above 2.5 (±150%) over the rest of the world (Table 1 and Fig. 2e). On local and monthly scales, the actual a priori errors for soil and biomass burning emissions might be higher than 3. However, because the top-down errors are generally smaller than a factor of 3 (section 3.4), our a posteriori will be dominated by the top-down constrains on soil and biomass burning emissions (see section 4), and thus a more detailed evaluation of a priori errors is not necessary for our analysis.
The a priori inventory is used to conduct a tropospheric O3–NOx–hydrocarbons simulation with the GEOS-CHEM model (version 5.05, http://www-as.harvard.edu/chemistry/trop/geos) for the year 2000. The model is driven by assimilated meteorological fields from the Goddard Earth Observing System (GEOS) Global Modeling and Assimilation Office, with a 2° latitude by 2.5° longitude horizontal resolution and 30 vertical levels. The resulting NO2 columns for four months (January, April, June and August 2000), chosen for illustrative purpose, are displayed on Fig. 3 (middle panels). These columns map onto surface NOx emissions because of the short lifetime of NOx (less than a day) and the large NO2/NOx ratio in the boundary layer.
 | ||
| Fig. 3 Tropospheric NO2 columns for January, April, June, and August 2000. Left panels: GOME observations. White areas indicate regions with no observations, or with cloud/snow cover >40%. Middle panels: GEOS-CHEM model results sampled at the GOME overpass time. Right panels: space-based observations of fires from VIRS20 on board TRMM for the tropics (latitudes equatorward of 40°) and ATSR21 on board ERS-2 at higher latitudes. All quantities are averaged over the GEOS-CHEM 2° × 2.5° horizontal grid. | ||
3. Top-down NOx emission inventory from GOME
3.1. The GOME instrument and retrieval
The GOME instrument is a nadir-viewing spectrometer, which detects absorption of atmospheric NO2 in the visible by measuring backscattered solar radiation. Global coverage is achieved in 3 d with a ground footprint of 320 km × 40 km (cross track × along track). Slant columns of NO2 are retrieved for the year 2000 by non-linear least-square fitting of backscattered radiance spectra.17 Our conversion from slant columns to tropospheric NO2 columns takes into account the influence of stratospheric columns and instrument biases, and includes scattering by the surface, clouds, aerosols, and gases.7,18We only consider scenes where clouds contribute to less than 50% of backscattered radiation, corresponding to cloud or snow cover <40%. The monthly mean uncertainties for a 2° × 2.5° grid box in the retrieved GOME tropospheric NO2 columns include a 30% relative error from the conversion of slant tropospheric columns to vertical tropospheric columns and a 5 × 1014 molecules cm−2 absolute uncertainty due to spectral fitting. A detailed error analysis is provided by Martin et al.18 Our relative error is slightly lower than the estimate given by Martin et al. because we have assumed random errors in spectral fitting, surface reflectivity and clouds. We do not use NO2 columns poleward of 60° N, where the retrieval is sometimes more uncertain.
3.2. GOME NO2 columns
The retrieved GOME tropospheric NO2 columns are compared to modeled columns in Fig. 3. Monthly mean observed and modeled NO2 columns are well correlated spatially and temporally (r = 0.75, n = 149477). Observations and model display enhanced NO2 columns over the industrialized regions of the eastern United States, Europe, India, East Asia, and South Africa throughout the year. Seasonal NOx emissions from large-scale tropical fires can also be seen during the dry seasons in North equatorial Africa (January), India and Southeast Asia (April), South equatorial Africa (June and August), and to a smaller extent over South America (August). Note the increased NO2 over Idaho and Montana in August 2000 due to unusually large wildfires.19 The spatial location of these seasonal enhancements matches space-based active fire observations (Fig. 3, right panels) from the Visible and Infrared Scanner (VIRS) on board the Tropical Rainfall Measuring Mission satellite (TRMM).20 The geographical coverage of VIRS fires spans tropical and subtropical latitudes (<40°). At higher latitudes, we supplement them with nighttime observations from the ATSR instrument21 on board ERS-2.During summer (Fig. 3, June and August), the GOME columns show regions of enhanced NO2 in areas not associated with fuel combustion or biomass burning: the western United States, southern Europe, the Middle East, the African Sahel, and parts of East Asia. These enhancements are not captured by the model, and are consistent with larger than expected NOx emissions from the seasonally dry soils in these regions.7,8
3.3. Global top-down NOx inventory
The methodology to infer surface NOx emissions from GOME NO2 columns is based on inverse modeling with GEOS-CHEM, as described by Martin et al.,7 linearly relating the observed NO2 columns to NOx emissions. The top-down emissions reflect surface NOx emissions: GOME columns over land are relatively insensitive to lightning NO emissions because of the low densities and low NO2/NO ratios in the upper troposphere.7,8 Recent studies have found evidence for lightning signatures in GOME NO2 columns over the Atlantic Ocean.22–24 Transient lightning signals were also observed over North America away from surface NOx sources.22The main source of uncertainty in this inversion comes from the ability of the model to accurately simulate the lifetime of NOx (i.e., to simulate the partitioning between NOx and total reactive nitrogen). This source of uncertainty was assessed to be less than 30%.7 The overall uncertainty in the top-down emission inventory is 42% (obtained by adding in quadrature a 30% error from the model inversion and a 30% error from the NO2 columns retrieval) which is combined with the 5 × 1014 molecules cm−2 absolute error. Our GOME inventory (with a global total of 40.5 TgN year−1, Table 1 and Fig. 2) underestimates actual top-down NOx emissions because of missing data caused by incomplete satellite sampling (for example over India) and areas with large cloud or snow cover (in particular during winter over eastern Europe, see Fig. 3). We assess this underestimate by sampling the a priori inventory only where GOME data is available, and find that 7% of total a priori emissions are thus missed. This observational undersampling will be corrected in the a posteriori inventory (section 4).
The top-down African emissions (6.9 TgN year−1) are 10% lower than our previously published results (7.8 TgN year−1), for the same GOME observations.8 The difference comes from using a new GEOS-CHEM simulation for the inversion analysis, with improved isoprene emissions matching the seasonality inferred by GOME HCHO observations over Africa (Jaeglé et al., manuscript in preparation).
3.4. Global partitioning of GOME top-down emissions
We derive the monthly partitioning of the top-down NOx emissions (E) among fuel combustion (EFF+BF), biomass burning (EBB) and soils (Esoil), by combining a priori information on the spatial location of areas dominated by fuel combustion, together with space-based observations of fires. The block diagram on Fig. 4 illustrates the two-step approach we apply to each month and each 2° × 2.5° grid box.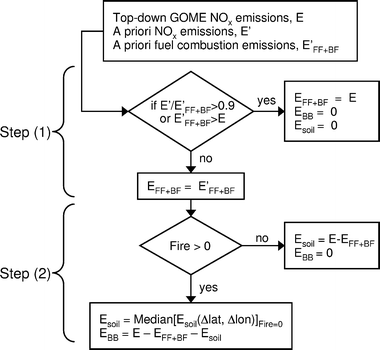 | ||
| Fig. 4 Block diagram of algorithm applied to partition top-down GOME NOx emissions (E) into: fuel combustion (EFF+BF), biomass burning (EBB) and soil (Esoil) emissions. The method is applied to monthly top-down NOx emissions in each 2° × 2.5° grid box, using information from the spatiotemporal distribution of bottom-up fuel combustion emissions (E′FF+BF) and space-based fire count observations (Fire). See text (section 3.4) for detailed description. | ||
For grid boxes where a priori total emissions (E′) are dominated by fuel combustion (E′FF+BF/E′ > 0.9) or where a priori combustion emissions exceed top-down emissions (E′FF+BF > E), we assume that all top-down emissions are from fuel combustion (EFF+BF = E, EBB = 0, Esoil = 0). Elsewhere (E′FF+BF/E′ ≤ 0.9 and E′FF+BF ≤ E) we set the top-down fuel combustion emissions to be equal to the a priori fuel combustion emissions (EFF+BF = E′FF+BF). This step thus takes advantage of the spatial separation between areas dominated by fuel combustion (northern hemisphere industrialized regions) and those dominated by soil or biomass burning (generally in the tropics).
Despite our use of the a priori in this first step, the large majority (80%) of the resulting GOME fuel combustion inventory is still determined by top-down emissions (EFF+BF = E). This is because fuel combustion emissions are concentrated over small geographical areas where they dominate overall emissions; these regions are identified by our E′FF+BF/E′ > 0.9 condition and account for 76% of global GOME fuel combustion emissions. Regions where a priori combustion emissions exceed top-down emissions (E′FF+BF > E) account for only 4% of GOME fuel combustion emissions and are located over SE Asia/India. The remaining 20% of top-down fuel combustion emissions are directly determined by the a priori (EFF+BF = E′FF+BF); these are mostly tropical regions with large soil or biomass burning emissions. We selected the E′FF+BF/E′ > 0.9 threshold by applying our algorithm to a synthetic inventory based on sampling the total a priori NOx inventory only when GOME observations are available. With this threshold we were able to retrieve the original monthly regional a priori fuel combustion emissions to better than 2%, while at the same time minimizing the error from neglecting soil and biomass burning emissions in industrialized regions, and maximizing the fraction of fuel combustion emissions determined by the top-down inventory. Using the synthetic inventory we estimate that 7% (0.7%) of soil (biomass burning) emissions are located in areas with E′FF+BF/E′ > 0.9. This underestimate of soil emissions is corrected in the optimal combination of the a priori and top-down emissions inventories in section 4. Varying the threshold from 0.85 to 0.95 has a small effect our top-down retrieval of fuel combustion (less than 5%).
In the second step, we use monthly fire observations (Fire) to separate biomass burning from soil emissions. If no fires are detected in a grid box for a given month, we assign the residual top-down emissions to soils (Esoil = E − EFF+BF, EBB = 0). For each grid box where fires are detected (Fire > 0), we need to separate background soil emissions from biomass burning emissions. From our a priori inventory, we estimate that ∼20–30% of emissions over biomass burning regions come from background soil emissions, which we thus need to take into account. This is achieved by calculating the median soil emissions in grid boxes within Δlat = 6° latitude and Δlon = 10° longitude where there are no fires:
| if Fire[x,y] > 0 then Esoil[x,y] = Median(Esoil[x′,y′]), for Fire[x′,y′] = 0 and |x′ − x| ≤ Δlon and |y′ − y| ≤ Δlat | (1) |
The nighttime fire detection of ATSR likely undersamples tropical fires, which are characterized by a marked diurnal cycle.25,26 In contrast, the orbit of TRMM permits the complete sampling of the diurnal burning cycle each month by VIRS.20 In addition, the VIRS retrieval algorithm takes into account multiple satellite overpasses and cloud masking. We thus use VIRS observations where available (in the tropics and subtropics), and elsewhere we use ATSR observations. We eliminate false detections in ATSR due to oil and gas flaring.27
In this second step, the separation between emissions from soils and biomass burning relies only on the spatial distribution of observed fires and not on their intensity. This is an important point as comparisons among different remotely sensed biomass burning datasets have shown good agreement in identifying burning regions, but large disagreement in terms of the magnitude of the areas affected.26,28–30 We test the sensitivity of our method to the choice of fire indicators by using ATSR everywhere or the Global Burned Area 2000 (GBA-2000) dataset from the SPOT VEGETATION sensor,31 and find that our results are affected by less than 10%.
As described above, the robustness of our algorithm is assessed by applying it to partition a synthetic inventory based on the total a priori surface NOx emissions. The original monthly regional a priori biomass burning and soil emissions are retrieved to within better than 10%, on average. We thus estimate relative errors in the individual top-down fuel combustion, soil, and biomass burning to be ∼45% (including a 42% error from the top-down retrieval, 10% error from the partitioning algorithm, and 10% error from fire detection), which add to the absolute error.
4. A posteriori NOx emissions
We combine the top-down NOx emission inventory, E and its relative error ε, with a priori estimates from the bottom-up inventory, E′ and its relative error ε′, to obtain an a posteriori inventory, E″. If we assume a log-normal distribution of errors, the optimized inventory is obtained as7 | (2) |
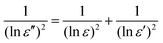 | (3) |
The a priori and top-down relative errors for total NOx emissions are generally comparable over source regions. The two inventories are thus given similar weights in the optimized inventory. The resulting a posteriori error is a factor of two lower compared to the a priori (Table 1 and Fig. 2). A similar reduction in the a posteriori error is found for the fuel combustion inventory. For soil and biomass burning emissions, the a priori relative errors (3) are much larger than the top-down errors (1.5–2), and thus the resulting a posteriori inventory is dominated by the top-down estimates of NOx emissions. However, over areas where GOME observations are not available or top-down emissions are equal to zero, the a posteriori directly reflects the bottom-up inventory. Thus over regions with a significant fraction of missing data, a posteriori errors can sometimes be larger than the top-down errors (Tables 3 and 4).
5. Results
5.1. Fuel combustion emissions
The top-down GOME fuel combustion inventory is well correlated with the a priori inventory (r = 0.85, n = 35160), as shown in Fig. 2b. The GOME inventory (24.6 TgN year−1) is 3% lower compared to the a priori (25.4 TgN year−1). Over the United States, Europe and East Asia the two inventories agree to within 10%, lending confidence to our inversion and partitioning algorithm (Fig. 5 and Table 2). More pronounced differences are noted elsewhere: the GOME inventory is larger than the a priori over Japan (50%) and S. Africa (45%), but lower over SE Asia/India (−45%), Central America (−20%), South America (−30%) and Australia (−40%).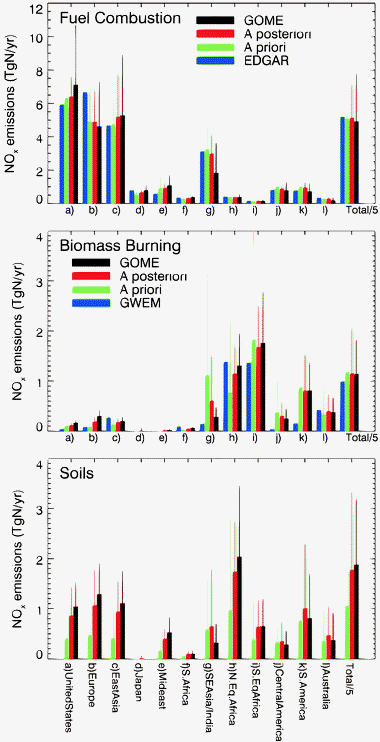 | ||
| Fig. 5 Regional GOME top-down (black), a priori (green), and a posteriori (red) annual NOx emissions in TgN year−1 for 2000: (top panel) fuel combustion emissions, (middle panel) biomass burning, and (bottom panel) soil emissions. The thin lines on top of each bar displays the absolute errors. Note that global emissions (rightmost bars) are divided by a factor of 5. Also shown are the inventories from EDGAR16 for fuel combustion (blue bars on top panel) and GWEM41 for biomass burning (blue bars on middle panel). The geographical definition of each region is displayed on Fig. 1. | ||
While the annual a posteriori and a priori fuel combustion emissions are very close to each other (25.6 TgN year−1 and 25.4 TgN year−1, respectively), information from GOME has helped reduce the global annual errors by a factor of 2 (from ±80% to ±40%). The a priori and a posteriori fuel combustion inventories are very well correlated (r = 0.96, n = 35 160). Regionally, the a posteriori is within 10% of the a priori, with the exception of Japan and S. Africa where the a posteriori is larger by 25%. Regions of disagreement where the top-down inventory was lower than the a priori were caused by under sampling by GOME (section 3.3) and disappear in the a posteriori. The fuel combustion inventory from EDGAR16 (26.1 TgN year−1) is also very similar to our a priori and a posteriori inventories (Table 2 and Fig. 5). The largest differences are for Europe and the Middle East, where EDGAR emissions are 37% larger and 33% lower than the a posteriori, respectively.
The monthly a posteriori fuel combustion emissions are aseasonal over most regions, with the exception of Europe and East Asia (Fig. 6, grey bars). Our a priori fuel combustion inventory does not include any seasonality over East Asia. However the a posteriori inventory suggests larger fuel combustion emissions during the winter months. The ratio of monthly emissions in December (maximum) to emissions in July (minimum) is 1.4. Streets et al.32 examined the potential seasonality of Chinese NOx emissions due to heating in homes, assuming a dependence of stove operation on outdoor temperature, and estimated a 1.2 ratio between maximum and minimum fuel combustion emissions. Their calculation neglected seasonality in power generation and industrial energy use, which are also likely to vary seasonally. Over Europe we derive a ratio between maximum and minimum emissions of 1.3, similar to seasonality in the a priori, which is due to wintertime heating.
 | ||
| Fig. 6 Monthly regional a posteriori (bars) and a priori (lines) NOx emissions in 2000. We show total emissions (light grey bars for a posteriori and black line for a priori) as well as their partitioning among fuel combustion (dark grey), biomass burning (red), and soil emissions (blue). Note the change in scales for the panels on each row. | ||
5.2. Biomass burning emissions
The global annual biomass burning inventories for the a priori (5.9 TgN year−1), top-down (5.7 TgN year−1) and a posteriori (5.8 TgN year−1) are nearly identical, but the inclusion of GOME observations greatly reduces errors, from ±200% to ±80%. There is good spatial and temporal correlation among these inventories (r = 0.69 for GOME vs. a priori; r = 0.85 for a posteriori vs. a priori, n = 35 160). Regionally the largest differences are for SE Asia/India, where the a posteriori (0.6 TgN year−1) is 46% lower than the a priori (1.1 TgN year−1), and for North equatorial Africa, where the a posteriori (1.2 TgN year−1) is 50% larger than the a priori (0.77 TgN year−1) (Table 3 and Fig. 5). For North equatorial Africa, the underestimate of the a priori is likely a result of an underestimate of the NO emission factor (EF = 1.1 (g NO) (kg dry matter)−1) used in our inventory for North African savannas.7,8 Over South equatorial Africa, where we use a higher EF (3.9 g kg−1),33 there is closer agreement between the a priori (1.8 TgN year−1) and the a posteriori (1.7 TgN year−1). Overall, fires over Africa account for half of the global biomass burning emissions. Globally, biomass burning emissions contribute 14% of surface NOx emissions.The a priori and a posteriori display similar seasonality over the major biomass burning regions, with maximum emissions in February for North equatorial Africa, March for Central America, April for SE Asia/India, July in South equatorial Africa, September in Australia and October in South America (Fig. 6, compare red bars and red lines). This good agreement on the seasonal cycle of burning validates the approach used by Duncan et al.12 to specify seasonal variations in the a priori, via regional scaling of a bottom-up inventory with fire count and aerosol satellite observations.
5.3. Soil emissions
Globally, our top-down soil emissions (9.4 TgN year−1) are 77% larger than the a priori (5.3 TgN year−1); the a posteriori soil emissions (8.9 TgN year−1) are 68% larger than the a priori. The spatial distribution of our inferred annual soil emissions corresponds well to the a priori (r = 0.51, n = 2349 for GOME vs. a priori; r = 0.79, n = 2741 for a posteriori vs. a priori). The large underestimate of soil emissions by the a priori appears to be systematic for all regions (see Fig. 5, bottom panel, and Table 4), and is especially strong during the summer over mid-latitude regions and during the wet season in the Tropics (Fig. 6).North equatorial Africa is identified as having large soil emissions (Fig. 2, Table 4). We previously argued that these NOx emissions are due to rain-induced pulsing from soils over vast areas of the Sahel.8 Many field and laboratory experiments have reported large pulses of biogenic NO emissions following rain on dry soils of savannas and seasonally dry forests.34–38 We found that the spatial and temporal distribution of the GOME NO2 enhancements over Africa is consistent with soil NO emissions from ecosystem-dependent flux measurements39 and from surface measurements of NO2 obtained by the IDAF (IGAC/DEBITS/Africa) network over West Africa. We showed that lightning and clouds are unlikely to account for the enhanced GOME NO2 columns and IDAF surface NO2 observations.
Here we see that the large inferred soil emissions are not restricted to the tropics, but extend to mid-latitude regions as well. In particular, our a posteriori analysis indicates a factor of two increase in soil emissions over Europe, the US and East Asia (Fig. 5, Table 4).
6. Comparison of a posteriori to previous studies
Table 1 compares our 2000 a posteriori inventory with the a posteriori inventory of Martin et al.,7 based on GOME observations for September 1996–August 1997. Our global a posteriori NOx source is 7% larger than their 37.8 TgN year−1a posteriori source. Most of the difference is for Tropical regions and could reflect interannual variations in biomass burning.Müller and Stavrakou40 used an adjoint modeling technique based on the IMAGES model to derive an optimized NOx emission inventory using GOME NO2 columns for 1997, as well as surface observations of CO. Their inversion resulted in a 38.7 TgN year−1 annual surface NOx emissions, which is close to our a posteriori estimate of 40.3 TgN year−1. Their global fuel combustion source (22.8 TgN year−1) and biomass burning source (4.4 TgN year−1) are 11% and 24% lower than our a posteriori, respectively. In agreement with our results, they infer a strong role for soil NOx emissions, with even larger global emissions (12.1 TgN year−1). Their NOx emissions over North America, Europe and South Asia (8.3, 5, 5.2 TgN year−1) are similar to our results (7.8, 4.9, 5.6 TgN year−1 – using the same regional definitions). However, they infer larger sources over S. America and Africa (6.1 and 8.8 TgN year−1) compared to our study (4.3 and 7 TgN year−1). While our study generally agrees with the results of Müller and Stavrakou,40 some of the discrepancies noted above are likely the results of differences in: a priori emissions (IMAGES uses larger a priori fuel combustion and soil NOx emissions), years, retrieval approaches for the GOME NO2 columns, and chemical mechanisms used in the inversions.
6.1. Biomass burning emissions
Table 3 and Fig. 5 compares our a posteriori biomass burning estimate with the Global Wildland Fire Emission Model 1.4 (GWEM) inventory of Hoelzemann.41 This inventory is an improvement of an earlier inventory (GWEM 1.21).42 It is based on the GBA-2000 burned area product (with the exception of S. America where GOES and AVHRR fire counts are used) and fuel loads from the LPJ-DGVM model. These inventories are in broad agreement, with the GWEM global inventory (5 TgN year−1) being 13% lower than our a posteriori (5.8 TgN year−1). We note that the GWEM inventory does not include seasonal emissions from burning of agricultural residues in the field, while our biomass burning inventories take this source into account. Open field agricultural residue burning produces 0.2 TgN year−1, as derived in the inventory of Logan and Yevich10 (which is used in our a priori), and is thus unlikely to fully account for the differences between GWEM and our inventory.Biomass burning estimates from GWEM and our a posteriori are lower than some of the earlier studies. For example the climatological emission inventory of Galanter et al.43 yields 7.8 TgN year−1. The Schultz27 inventory for 2000 yields 7.5 TgN year−1.
Both GWEM and a posteriori estimates display reduced emissions over SE Asia/India relative to the a priori, by 86% and 46%, respectively. Similar conclusions were reached by previous studies based on inverse modeling analyses of CO observations.44,45 Our a posteriori estimates for biomass burning emissions of NOx over Asia (0.8 TgN year−1) agrees with estimates from a recent bottom-up inventory by Streets et al.46 (0.85 TgN year−1). The convergence towards lower biomass burning emissions over Asia is likely to be due to an overestimate in the amount of biomass burnt used in early studies, especially over India.46
For the African continent (south of the Sahara), the GWEM inventory and our a posteriori both estimate 2.9 TgN year−1 for NOx emissions. However, while in GWEM African emissions north and south of the equator are nearly equal, in our a posteriori we find that burning south of the equator is 40% larger than north of the equator. For comparison, Barbosa et al.47 estimate NOx emissions from vegetation fires of 1.2–4.5 TgN year−1 over Africa during the 1980s, while Scholes and Andreae48 find 2.8 TgN year−1, similar to our estimates.
The largest difference between GWEM and our a posteriori occurs over South and Central America, which account for 0.2 TgN year−1 in GWEM compared to 1.1 TgN year−1 in the a posteriori. Our a posteriori contains large uncertainties over this region because of the South Atlantic anomaly.49 However, burned areas in South America are probably underestimated in GWEM (J. Hoelzemann, personal communication), because with its 4 km resolution the GOES fire product might not detect many tropical deforestation fires, which typically cover small areas (0.2–1 km2).50 In comparison, Potter et al.51 find that deforestation fires over the Amazon in the early 1990s resulted in 1.6 TgN year−1.
In addition to the GWEM inventory, two other global studies (van der Werf et al.52 and Ito and Penner53) have estimated trace gas emissions from biomass burning during 2000 based on remote sensing of burned areas and active fires. NOx emissions have not been reported from these other two inventories. However, comparisons of the carbon emissions for these three studies show significant differences,28 with global emissions ranging from 1428 TgC to 2600 TgC, African emissions ranging from 880 TgC to 1880 TgC, and South/Central American emissions ranging from 90 TgC year−1 to 760 TgC year−1. This range reflects highly different estimates of both burned areas29,54 and available fuel loads.28 Given such difficulties in relating space-based observations of active fires and burned areas to gas emissions, we propose that our a posteriori biomass burning inventory could provide a way to independently validate the algorithms used in these recent studies for the year 2000.
6.2. Asian fuel combustion emissions
Our a posteriori estimate for fuel combustion over Asia (8.8 TgN year−1, including East Asia, SE Asia/India and Japan) is 28% larger than the bottom-up inventory of Streets et al.32 (6.9 TgN year−1). The largest discrepancies are for China and India, where our fuel combustion emissions (4.4 TgN year−1 and 1.7 TgN year−1) are 38% and 43% higher than the Streets et al. inventory, respectively. Modeling analyses based on CO observations by aircraft and satellite observations during the TRACE-P field mission44,45,55,56 also found that the Streets et al. inventory significantly underestimates anthropogenic emissions over China. Causes for this underestimate could include under-reporting of domestic coal burning32,56 or inefficient industrial sources and power plants.57In an inverse modeling study using NOyin situ observations, Wang et al.58 inferred a 47% increase in Chinese NOx emissions, relative to the Streets et al. inventory. They estimate a 5 TgN year−1 total NOx source for China, which is close to our total a posteriori source (5.3 TgN year−1).
6.3. Soil emissions
The global magnitude of soil-atmospheric NOx exchange has been challenging to constrain because of the large spatiotemporal variability of microbial soil processes,59,60 the influence of human activities61 such as N-fertilization62 and burning37 in enhancing soil emissions, and the role of plant canopies in recapturing a significant fraction of the emitted soil NOx.63,64 The most recent global soil inventories include the semi-empirical model of Yienger and Levy14 (used in our a priori), the process-based land-vegetation biogeochemistry model of Potter et al.,65 and the inventory of Davidson and Kingerlee66 based on global extrapolations of surface flux measurements. The above-canopy soil emissions derived by Yienger and Levy and Potter et al. (5.5 and 5 TgN year−1, respectively) are a factor of two lower than the results of Davidson and Kingerlee (13 TgN year−1). The global soil NOx estimate from our a posteriori, with 8.9 TgN year−1, is in between these two values. By construction, our a posteriori estimate depends on the a priori. We conducted a sensitivity study where we doubled the a priori soil emissions, and derived an a posteriori value of 10.4 TgN year−1 for soil emissions, closer to values obtained by Davidson and Kingerlee and by Müller and Stavrakou40 in their inversion analysis.The largest biogenic emissions of NOx have been measured from fertilized agriculture and seasonally dry tropical ecosystems.66 These systems support rapid nitrogen cycling (via nitrification and denitrification), high temperatures, and/or seasonally low moisture. Consistent with these field measurements, our study highlights the role of tropical savanna/woodland ecosystems as a large source of NOx. We also find large emissions from mid-latitude grassland and cropland areas during summer, especially over the western US (Great Plains), southern Europe (Spain, Greece, Turkey), China, and India (Fig. 2).
Africa accounts for nearly 30% of our global a posteriori soil emissions. These emissions peak at the beginning of the rainy season, in June (Fig. 6h). The difference between our a posteriori and the a priori during the rainy season suggests that the Yienger and Levy algorithm underestimates the magnitude of rain-induced pulsing.8
Relative to the a priori inventory, we find a larger increase in northern mid-latitude soil emissions (25° N–60° N: 1.7 TgN year−1a priori, compared to 3.9 TgN year−1 in a posteriori) than in the tropical emissions (25° S–25° N: 3 TgN year−1a priori, compared to 4.3 TgN year−1 in a posteriori). This larger role for mid-latitude emissions could reflect a strong source of NOx from fertilized agricultural soils. Global estimates of soil emissions from agricultural fields and the effect of nitrogen fertilization vary widely, ranging from 0.5 to 3 TgN year−114,62,67,68 (not including canopy recapture). Including the canopy recapture of Yienger and Levy, these numbers range from 0.4 to 2.25 TgN year−1. The main uncertainty in these estimates comes from assessing the fraction of applied N-fertilizer released as NO, which vary from 0.3%62 at the low end, to 2.5%14 at the high end of these compilations. By multiplying the area of cropland by an average annual emission flux based on observations, Davidson and Kingerlee estimate an even larger source of 5.4 TgN year−1 (3.9 TgN year−1 with canopy recapture).
We estimate the role of emissions from agricultural fields by scaling our a posteriori soil emissions by the fraction of emissions coming from agriculture. Yienger and Levy find that 75% of emissions from temperate soils and 25% of emissions from tropical soils come from agriculture, while in Davidson and Kingerlee these numbers are 21% and 28%. We thus find that, globally, 2.5–4.5 TgN year−1 are emitted by N-fertilized soils: with 1.1–1.6 TgN year−1 in the Tropics and 0.9–3.4 TgN year−1 in the extratropics. These numbers are at the upper end of the previous estimates listed above. Asia contains one third of the world’s total cropland area and accounts for more than half of the global N-fertilizer consumption,69 with China and India being the largest users. Indeed, our a posteriori soil emissions are highest over the North China Plain and North India (Fig. 2), which are regions with extensive croplands and where chemical N-fertilizers are heavily used. In their inversion study for Asia, Wang et al.58 proposed that decomposition of organic wastes and extensive applications of chemical fertilizer could account for 1.1 TgN year−1 over China alone. This is consistent with our a posteriori estimate of ∼1 TgN year−1 from soil emissions over East Asia. In contrast, by using a bottom-up approach, Yan et al.67 estimate that only 0.2 TgN year−1 are emitted from croplands in China. These discrepancies clearly warrant a close re-examination of fertilizer-induced NOx release.
Globally, soil emissions account for 22% of NOx emissions in our a posteriori inventory, but only 14% of the a priori. The a posteriori indicates that in the Tropics, soil emissions (4.3 TgN year−1) are comparable to biomass burning emissions (4.7 TgN year−1), while in the a priori soil emissions (3 TgN year−1) are 40% lower than biomass burning (5 TgN year−1). In addition, we infer a large role for soil emissions at northern mid-latitudes during summer (June–August), where they account for nearly half that of the fuel combustion source (compared to only 20% in the a priori). This suggests that the current generation of global and regional chemical transport models (which generally use low estimates of soil emissions) likely underestimate the role of biogenic emissions in controlling background levels of ozone, especially during the rainy season in the Tropics and during summer for northern mid-latitudes.
7. Conclusions
We have presented a simple method to partition global NOx emissions derived from the Global Ozone Monitoring Experiment (GOME) into fuel combustion, biomass burning and soil emissions for the year 2000. Our resulting a posteriori fuel combustion emissions (25.6 TgN year−1) are very close to the GEIA-based a priori (25.4 TgN year−1), lending confidence to our separation method, and errors are reduced by a factor of two from ±80% to ±40%. Fuel combustion emissions are aseasonal, with the exception of Europe and East Asia.The global a posteriori estimate of biomass burning emissions (5.8 TgN year−1) is similar to the a priori (5.9 TgN year−1), with a significant decrease in the uncertainty from ±200 to ±80%. The a posteriori estimate of NOx emissions from SE Asia/India is decreased by 46% relative to the a priori, similar to the results of previous studies based on inverse modeling analyses of CO observations. Over North equatorial Africa, a posteriori emissions are 50% larger than the a priori, likely due to errors in the NOx emission factor used in the a priori. We propose that our a posteriori biomass burning inventory can be used to validate independent bottom-up estimates of fire emissions based on remote sensing of burned areas and active fires for 2000.
We find a 68% increase in soil emissions from 5.3 TgN year−1 to 8.9 TgN year−1. The a posteriori inventory displays the largest soil emissions over tropical savanna/woodland ecosystems during the wet season, as well as over agricultural regions in mid-latitudes during summer. We estimate that 2.5–4.5 TgN year−1 are emitted by N-fertilized soils, at the upper end of previous estimates. The large underestimate of atmospheric-soil NOx exchange by the Yienger and Levy14 algorithm used in our a priori, appears to reflect an underestimate of N-fertilized NO release from croplands as well as an underestimate of rain-induced pulsing from semiarid soils. This points to a clear need to better constrain these two effects, and re-evaluate the role of soil emissions in controlling background ozone levels.
Acknowledgements
This work was supported by funding from the NASA New Investigator Program in Earth Science (NAG5-10637). The GEOS-CHEM model is managed by the Atmospheric Chemistry Modeling group at Harvard University with support from the NASA Atmospheric Chemistry Modeling and Analysis Program. We thank J. Hoelzemann for providing us with the gridded GWEM 1.4 inventory.References
- M. J. Prather and D. Ehhalt, in Climate Change 2001: The Science of Climate Change, Intergovernmental Panel on Climate Change, Cambridge University Press, Cambridge, 2001, p. 241 Search PubMed.
- J. A. Logan, J. Geophys. Res., 1983, 88, 785 CrossRef.
- E. A. Holland, F. J. Dentener, B. H. Braswell and J. M. Sulzman, Biogeochemistry, 1999, 46, 7 CAS.
- ESA, The GOME users manual, ESTEC, European Space Agency Publication SP-1182, ESA Publications Division, Noordwijk, The Netherlands, 1995, ISBN-92-9092-327-x Search PubMed.
- J. P. Burrows, M. Weber, M. Buchwitz, V. Rozanov, A. Ladstatter-Weissenmayer, A. Richter, R. DeBeek, R. Hoogen, K. Bramstedt, K. U. Eichmann and M. Eisinger, J. Atmos. Sci., 1999, 56, 151 CrossRef.
- C. Leue, M. Wenig, T. Wagner, O. Klimm, U. Platt and B. Jahne, J. Geophys. Res., 2001, 106, 5493 CrossRef CAS.
- R. V. Martin, D. J. Jacob, K. Chance, T. P. Kurosu, P. I. Palmer and M. J. Evans, J. Geophys. Res., 2003, 108 DOI:10.1029/2003JD003453.
- L. Jaeglé, R. V. Martin, K. Chance, L. Steinberger, T. P. Kurosu, D. J. Jacob, A. I. Modi, V. Yoboué, L. Sigha-Nkamdjou and C. Galy-Lacaux, J. Geophys. Res., 2004, 109 DOI:10.1029/2004JD004787.
- I. Bey, D. J. Jacob, R. M. Yantosca, J. A. Logan, B. D. Field, A. M. Fiore, Q. B. Li, H. G. Y. Liu, L. J. Mickley and M. G. Schultz, J. Geophys. Res., 2001, 106, 23073 CrossRef CAS.
- R. Yevich and J. A. Logan, Global Biogeochem. Cycles, 2003, 17 DOI:10.1029/2002GB001952.
- J. M. Lobert, W. C. Keene, J. A. Logan and R. Yevich, J. Geophys. Res., 1999, 104, 8373 CrossRef CAS.
- B. N. Duncan, R. V. Martin, A. C. Staudt, R. Yevich and J. A. Logan, J. Geophys. Res., 2003, 108 DOI:10.1029/2002JD002378.
- C. M. Benkovitz, M. T. Scholtz, J. Pacyna, L. Tarrason, J. Dignon, E. C. Voldner, P. A. Spiro, J. A. Logan and T. E. Graedel, J. Geophys. Res., 1996, 101, 29239 CrossRef CAS.
- J. J. Yienger and H. Levy, J. Geophys. Res., 1995, 100, 11447 CrossRef CAS.
- Y. H. Wang, D. J. Jacob and J. A. Logan, J. Geophys. Res., 1998, 103, 10713 CrossRef CAS.
- J. G. J. Olivier and J. J. M. Berdowski, in The Climate System, ed. J. Berdowski, R. Guicherit and B.J. Heij, AA Balkema Publishers, Lisse, The Netherlands, 2001, p. 33 Search PubMed.
- K. Chance, P. I. Palmer, R. J. D. Spurr, R. V. Martin, T. P. Kurosu and D. J. Jacob, Geophys. Res. Lett., 2000, 27, 3461 CrossRef CAS.
- R. V. Martin, K. Chance, D. J. Jacob, T. P. Kurosu, R. J. D. Spurr, E. Bucsela, J. F. Gleason, P. I. Palmer, I. Bey, A. M. Fiore, Q. B. Li, R. M. Yantosca and R. B. A. Koelemeijer, J. Geophys. Res., 2002, 107 DOI:10.1029/2001JD002622.
- J. F. Lamarque, D. P. Edwards, L. K. Emmons, J. C. Gille, O. Wilhelmi, C. Gerbig, D. Prevedel, M. N. Deeter, J. Warner, D. C. Ziskin, B. Khattatov, G. L. Francis, V. Yudin, S. Ho, D. Mao, J. Chen and J. R. Drummond, Geophys. Res. Lett., 2003, 30 DOI:10.1029/2003GL017503.
- L. Giglio, J. D. Kendall and R. Mack, Int. J. Remote Sens., 2003, 24, 4505 CrossRef.
- O. Arino and J. M. Melinotte, Int. J. Remote Sens., 1998, 19, 2019 CrossRef.
- Y. Choi, Y. H. Wang, T. Zeng, R. V. Martin, T. P. Kurosu and K. Chance, Geophys. Res. Lett., 2005 Search PubMed , in press.
- A. Richter and J. P. Burrows, Adv. Space Res., 2002, 29, 1673 CrossRef CAS.
- D. P. Edwards, J. F. Lamarque, J. L. Attie, L. K. Emmons, A. Richter, J. P. Cammas, J. C. Gille, G. L. Francis, M. N. Deeter, J. Warner, D. C. Ziskin, L. V. Lyjak, J. R. Drummond and J. P. Burrows, J. Geophys. Res., 2003, 108 DOI:10.1029/2002JD002927.
- S. Langaas, Nature, 1993, 363, 120 CrossRef.
- H. Eva and E. F. Lambin, Remote Sens. Environ., 1998, 64, 292 CrossRef.
- M. G. Schultz, Atmos. Chem. Phys., 2002, 2, 387 Search PubMed.
- E. S. Kasischke and J. E. Penner, J. Geophys. Res., 2004, 109 Search PubMed , doi:10.1029/2004JD004972.
- L. Boschetti, H. D. Eva, P. A. Brivio and J. M. Grégoire, Geophys. Res. Lett., 2004, 31 DOI:10.1029/2004GL021229.
- M. Simon, S. Plummer, F. Fierens, J. J. Hoelzemann and O. Arino, J. Geophys. Res., 2004, 109 DOI:10.1029/2003JD003622.
- J. M. Grégoire, K. Tansey and J. M. N. Silva, Int. J. Remote Sens., 2003, 24, 1369 CrossRef.
- D. G. Streets, T. C. Bond, G. R. Carmichael, S. D. Fernandes, Q. Fu, D. He, Z. Klimont, S. M. Nelson, N. Y. Tsai, M. Q. Wang, J. H. Woo and K. F. Yarber, J. Geophys. Res., 2003, 108 DOI:10.1029/2002JD003093.
- M. O. Andreae and P. Merlet, Global Biogeochem. Cycles, 2001, 15, 955 Search PubMed.
- C. Johansson and E. Sanhueza, J. Geophys. Res., 1988, 93, 14193 CrossRef CAS.
- E. A. Davidson, Soil Sci. Soc. Am. J., 1992, 56, 95 CAS.
- G. W. Harris, F. G. Wienhold and T. Zenker, J. Geophys. Res., 1996, 101, 23707 CrossRef CAS.
- J. S. Levine, E. L. Winstead, D. A. B. Parsons, M. C. Scholes, R. J. Scholes, W. R. Cofer, D. R. Cahoon and D. I. Sebacher, J. Geophys. Res., 1996, 101, 23689 CrossRef CAS.
- M. C. Scholes, R. Martin, R. J. Scholes, D. Parsons and E. Winstead, Nutr. Cycl. Agroecosyst., 1997, 48, 115 Search PubMed.
- D. Serça, R. Delmas, X. Le Roux, D. A. B. Parsons, M. C. Scholes, L. Abbadie, R. Lensi, O. Ronce and L. Labroue, Global Biogeochem. Cycles, 1998, 12, 637 CrossRef CAS.
- J. F. Müller and T. Stavrakou, Atmos. Chem. Phys. Disc., 2005, 4, 7985 Search PubMed.
- J. J. Hoelzemann, Global wildland fire emission modeling: Assessing variability of emissions and their impact on atmospheric chemistry, PhD thesis, Max Planck Institute for Meteorology, Hamburg, 2005 Search PubMed.
- J. J. Hoelzemann, M. G. Schultz, G. P. Brasseur, C. Granier and M. Simon, J. Geophys. Res., 2004, 109 DOI:10.1029/2003JD003666.
- M. Galanter, H. Levy and G. R. Carmichael, J. Geophys. Res., 2000, 105, 6633 CrossRef CAS.
- P. I. Palmer, D. J. Jacob, D. B. A. Jones, C. L. Heald, R. M. Yantosca, J. A. Logan, G. W. Sachse and D. G. Streets, J. Geophys. Res., 2003, 108 DOI:10.1029/2003JD003397.
- C. L. Heald, D. J. Jacob, D. B. A. Jones, P. I. Palmer, J. A. Logan, D. G. Streets, G. W. Sachse, J. C. Gille, R. N. Hoffman and T. Nehrkorn, J. Geophys. Res., 2004, 109 DOI:10.1029/2004JD005185.
- D. G. Streets, K. F. Yarber, J. H. Woo and G. R. Carmichael, Global Biogeochem. Cycles, 2003, 17 Search PubMed , doi:10.1029/2003GB002040.
- P. M. Barbosa, D. Stroppiana, J. M. Gregoire and J. M. C. Pereira, Global Biogeochem. Cycles, 1999, 13, 933 CrossRef CAS.
- M. Scholes and M. O. Andreae, Ambio, 2000, 29, 23.
- J. R. Heirtzler, J. Atmos. Sol. Terr. Phys., 2002, 64, 1701 CrossRef.
- Y. J. Kaufman, P. V. Hobbs, V. Kirchhoff, P. Artaxo, L. A. Remer, B. N. Holben, M. D. King, D. E. Ward, E. M. Prins, K. M. Longo, L. F. Mattos, C. A. Nobre, J. D. Spinhirne, Q. Ji, A. M. Thompson, J. F. Gleason, S. A. Christopher and S. C. Tsay, J. Geophys. Res., 1998, 103, 31783 CrossRef CAS.
- C. Potter, V. Brooks-Genovese, S. Klooster and A. Torregrosa, J. Geophys. Res., 2002, 107 DOI:10.1029/2000JD000250.
- G. R. van der Werf, J. T. Randerson, G. J. Collatz, L. Giglio, P. S. Kasibhatla, A. F. Arellano, S. C. Olsen and E. S. Kasischke, Science, 2004, 303, 73 CrossRef CAS.
- A. Ito and J. E. Penner, J. Geophys. Res., 2004, 109 DOI:10.1029/2003JD004423.
- K. Tansey, J. M. Grégoire, D. Stroppiana, A. Sousa, J. Silva, J. M. C. Pereira, L. Boschetti, M. Maggi, P. A. Brivio, R. Fraser, S. Flasse, D. Ershov, E. Binaghi, D. Graetz and P. Peduzzi, J. Geophys. Res., 2004, 109 DOI:10.1029/2003JD003598.
- D. Allen and K. Pickering, and M. Fox-Rabinovitz, J. Geophys. Res., 2004, 109 DOI:10.1029/2003JD004250.
- G. R. Carmichael, Y. Tang, G. Kurata, I. Uno, D. G. Streets, N. Thongboonchoo, J. H. Woo, S. Guttikunda, A. White, T. Wang, D. R. Blake, E. Atlas, A. Fried, B. Potter, M. A. Avery, G. W. Sachse, S. T. Sandholm, Y. Kondo, R. W. Talbot, A. Bandy, D. Thorton and A. D. Clarke, J. Geophys. Res., 2003, 108 DOI:10.1029/2002JD003116.
- P. Suntharalingam, D. J. Jacob, P. I. Palmer, J. A. Logan, R. M. Yantosca, Y. P. Xiao, M. J. Evans, D. G. Streets, S. L. Vay and G. W. Sachse, J. Geophys. Res., 2004, 109 DOI:10.1029/2003JD004362.
- Y. X. Wang, M. B. McElroy, T. Wang and P. I. Palmer, J. Geophys. Res., 2004, 109 DOI:10.1029/2004JD005250.
- G. L. Hutchinson, M. F. Vigil, J. W. Doran and A. Kessavalou, Nutr. Cycl. Agroecosyst., 1997, 48, 25 Search PubMed.
- J. Ludwig, F. X. Meixner, B. Vogel and J. Forstner, Biogeochemistry, 2001, 52, 225 CrossRef CAS.
- E. Sanhueza, Nutr. Cycl. Agroecosyst., 1997, 48, 61 Search PubMed.
- U. Skiba, D. Fowler and K. A. Smith, Nutr. Cycl. Agroecosyst., 1997, 48, 139 Search PubMed.
- D. J. Jacob and P. S. Bakwin, in Microbial Production and Consumption of Greenhouse Gases: Methane, Nitrogen oxides, and Halomethanes, ed. J. E. Rogers and W. B. Whitman, American Society for Microbiology, Washington DC, 1991, p. 237 Search PubMed.
- L. N. Ganzeveld, J. Lelieveld, F. J. Dentener, M. C. Krol, A. J. Bouwman and G. J. Roelofs, J. Geophys. Res., 2002, 107 DOI:10.1029/2001JD001289.
- C. S. Potter, P. A. Matson, P. M. Vitousek and E. A. Davidson, J. Geophys. Res., 1996, 101, 1361 CrossRef CAS.
- E. A. Davidson and W. Kingerlee, Nutr. Cycl. Agroecosyst., 1997, 48, 37 Search PubMed.
- X. Y. Yan, H. Akimoto and T. Ohara, Global Change Biol., 2003, 9, 1080 Search PubMed.
- A. F. Bouwman, L. J. M. Boumans and N. H. Batjes, Global Biogeochem. Cycles, 2002, 16 DOI:10.1029/2001GB001812.
- FAO, Current world fertilizer trends and outlook to 2008/09, Food and Agriculture Organization of the United Nations, Rome, 2004 Search PubMed.
| This journal is © The Royal Society of Chemistry 2005 |