Control of pore hydrophilicity in ordered nanoporous polystyrene using an AB/AC block copolymer blending strategy
Huiming
Mao
,
Pedro L.
Arrechea
,
Travis S.
Bailey
,
Bret J. S.
Johnson†
and
Marc A.
Hillmyer
*
Department of Chemistry, University of Minnesota, 207 Pleasant St. SE, Minneapolis, Minnesota 55455-0431, USA. E-mail: hillmyer@chem.umn.edu
First published on 19th August 2004
Abstract
Ordered nanoporous plastics with hydrophilic pore surfaces were prepared by the degradative removal of polylactide from a self-organised, multi-component composite containing two block copolymers: polystyrene-polylactide and polystyrene-polyethylene oxide. The solid-state characterization of blends containing up to 12 wt.% polyethylene oxide was consistent with nanoscopic cylinders of mixed polyethylene oxide and polylactide hexagonally packed in a polystyrene matrix. Orientation of these materials through simple channel die processing resulted in good cylinder alignment. Subsequent methanolysis/hydrolysis of the polylactide component gave nanoporous polystyrene with polyethylene oxide coated pores. The resulting nanoporous materials were able to imbibe water, in contrast to nanoporous polystyrene with no polyethylene oxide component.
Introduction
Block copolymers are a class of self-organising polymers that have utility in a wide variety of applications: from asphalt modifiers1 to nanolithographic templates for magnetic storage media applications.2 As demonstrated in the latter application, the evolution of nanotechnology has embraced soft material self-assembly3 as a legitimate fabrication tool,4,5 and block copolymers certainly represent a class of materials that exhibit unparalleled versatility in this regard.6 Nanoporous materials derived from self-assembled block copolymers7 are a specifc class of materials with many possible nanotechnological applications, for example, nanoscopic templates8,9 or nanostructured separation media.10 One can also envision these nanoporous materials as high surface area supports for catalysis applications11 or as materials that confine chemical and physical processes on a nanometer length scale.12 Since the first report of a nanoporous material from an ordered block copolymer precursor by Nakahama in 1988,13 many groups have focused on the design, preparation, and applications of this class of versatile nanoporous materials.8,10,14–26The need for nanoporous materials with predictable and reproducible pore size, alignment, distribution, and geometry is based on the critical nature of these morphological features to the efficacy of any application that involves their use (e.g., separations,27 photonic materials,28 templates29,30). Furthermore, the precise pore wall functionality can be important in applications that require specific interactions between some substrate (or analyte) and the pore wall. Therefore the controlled introduction of a specific chemical functionality within the pore space can be equally as critical to a material's designed purpose. While a great deal of attention has been directed at chemical functionalization of inorganic nanoporous silica phases, the introduction of functional groups must often be accomplished through modification of residual silanol groups following calcination, or through complex co-condensation reactions complicated by solution phase removal of the templating phase; a process often destructive to the nanoporous structure itself.31 In contrast, the formation of polymeric nanoporous materials, based on block copolymer self-organisation in the melt state, can be designed to avoid chemically and thermally aggressive processing. As a result, the opportunities for functionalization in these systems can span the entire fabrication process, from the initial synthesis of the block copolymer to modification of the final nanoporous structure.
Nanoporous polymeric materials from ordered block copolymers are typically prepared by selective removal of a minority block from one of the traditional non-lamellar diblock copolymer phases, namely the sphere, gyroid, and cylinder morphologies. Through deliberate block selection (defining the Flory–Huggins interaction parameter, χ) and careful variation of the overall degree of polymerization (N) and composition (fA, fB, etc.), remarkable control over pore size, distribution, and geometry are afforded.32 Furthermore, block copolymer morphologies have proven conveniently susceptible to macroscopic alignment both in monolithic and thin film environments, giving rise to highly ordered materials at length scales far beyond typical pore dimensions. With the recent evolution of “living” and controlled polymerization techniques, the number of monomer types that can be incorporated into low polydispersity block copolymers has been increased considerably. Combined with the flexibility of these systems to tolerate controlled chemical modification before or after formation of the porous solid, block copolymers systems provide an extremely facile and tunable route to functional ordered nanoporous materials.
Several example strategies for the functionalization of polymeric nanoporous materials are depicted in Fig. 1. The most basic strategy exploits the incorporation of a specific chemical functionality at the covalent junction between the matrix and sacrificial blocks. In this arrangement, removal or degradation of the sacrificial block conveniently exposes the chemical functionality at the pore wall. This strategy has been demonstrated by Zalusky et al.33,34 and Wolf and Hillmyer35 in which the degradation of a sacrificial polylactide (PLA) block generates hydroxyl functional groups at the pore surfaces in polystyrene (PS) and polycyclohexylethylene (PCHE) monoliths, respectively. This strategy is limited by the intrinsic maximum areal density of functional groups defined as the interfacial area per chain. Furthermore, the functional groups need to be accessible (i.e., not buried as the result of surface reconstruction within the pores). In principle, moderate reduction of this intrinsic functional group density can be achieved through blending of non-functional matrix homopolymer, thus increasing the interfacial area associated with each functional group lining the pore wall.36,37
 | ||
| Fig. 1 Example routes to nanoporous materials with controlled pore-wall functionality. (a) A functional group is incorporated into the junction between matrix and sacrificial blocks, and is exposed upon template degradation. (b) A functionalized mid-block is inserted between the matrix and sacrificial end block, producing a functional polymer brush at the pore wall upon removal of the template. (c) An AB/AC diblock copolymer blend is formed in which the common A block serves as the matrix, the B and C blocks are miscible, and only one of the two blocks is susceptible to degradative removal. In this manner a functionalized non-degradable block can be introduced as a diffuse brush along the pore interior. | ||
One method of circumventing this natural limitation in functional group density is to introduce a third block intermediate between the matrix and templating blocks, as depicted in Fig. 1(b). By incorporating blocks with pre-existing functionality, (e.g., polyisoprene (PI) or polyvinylpyridine (PVP)) the brush-like arrangement of chains left following template removal generates a high density halo of functional groups along the pore's circumference. Control of the length of this middle block gives access to a wide range of functional group densities, given the volume fraction of this block is consistent with the desired morphology of the nanoporous material. Review of several references detailing the phase behavior of ABC triblock copolymer systems shows cylindrical and network morphologies consistent with desirable nanoporous structures, exist over a wide range of mid-block compositions.38–40 We are currently exploring this approach using core-shell cylinder forming PS-PI-PLA, PI-PS-PLA and PS-PVP-polycaprolactone triblock copolymer systems,41 and Liu et al. have described a related methodology.42
A final strategy, and the subject of this manuscript, is the ability to add functionality within the pore structure through the blending of AB and AC diblock copolymers. In this scenario, the A blocks common to both molecules form the matrix, the B and C blocks are miscible and form a composite templating phase, and only one of the two composite phase blocks (e.g., B) is susceptible to the degradation process (Fig. 1(c)). The resultant material thus contains a disperse brush of the non-degradable C block confined within the porous structure. The brush density and overall C block composition in the pore space is defined by the degree of polymerization of the C block and the overall composition of the blend. The overall functional group density in the pore can then be defined through control of the intra-chain density of functional groups in the brush as discussed in the second approach above. Provided block copolymers containing appropriate miscible B and C blocks can be prepared, this strategy is highly desirable, enabling systematic tuning of the above features.
As we will show in this report, the incorporation of a simple polyether into the pore space employing this method dramatically impacts the wettability of the resulting nanoporous material. Using this method, we can in principle generate monoliths capable of supporting catalysis, ion transport, and molecular separations in aqueous and biological media. Exploiting the partial miscibility of polyethylene oxide (PEO) and PLA,43–53 we employed a blend of PS-PLA33 and PS-PEO54 diblock copolymers to generate a single-phase cylindrical morphology in which the PEO and PLA blocks form composite cylinders within a PS matrix. While the phase behavior of a few AB/AC diblock copolymer blends have been reported,55–58 we are aware of only one report describing the formation of a single ordered morphology in which two of the components (B and C) completely mix to form a single domain (spherical or lamellar in this case).59
The parent blends and resultant nanoporous monoliths described here were examined using a collection of experimental techniques, revealing ordered materials with flow field induced pore anisotropy, narrow pore size distribution, and hydrophilic character completely absent in the unblended materials. The pore size and hydrophilic properties of these materials were easily adjusted through selection of the two component diblock copolymers and variation of their blending ratio. The hydrophilic nature of the pore walls imparted by this blending technique was demonstrated by absorption of water and observable density change and also by the diffusion of water-soluble dye and associated color change. Hydrophilicity was found to be exclusive to the PEO containing nanoporous monoliths.
Results and discussion
Preparation and characterization of parent PS-PLA and PS-PEO block copolymers
Four block copolymers were prepared for this study using established methods. Two PS-PLA copolymers and two PS-PEO copolymers were synthesized by anionic polymerization of styrene followed by end capping with ethylene oxide. The macroinitiators were used for the subsequent controlled “coordination-insertion” polymerization of D,L-lactide33 or anionic polymerization of ethylene oxide,60,61 to give model materials. The PLA component in the PS-PLA materials was atactic and completely amorphous. Molecular characterization of the four samples is given in Table 1. All samples exhibited narrow molecular weight distributions. The two PS-PLA copolymers (SL1 and SL2) contained approximately 30 vol% PLA and the two PS-PEO (SO1 and SO2) materials were nearly symmetric.| Samples | M n/kg mol−1 PSa | M n/kg mol−1 PLA (or PEO)b | M n/kg mol−1 total | M n/kg mol−1 totalc | M w/Mnc | f PLA (or fPEO)d | D/nme |
|---|---|---|---|---|---|---|---|
a Determined by SEC vs. PS standards. These values are in agreement with the 1H NMR end group analysis.
b Determined by 1H NMR spectroscopy.
c Determined by SEC.
d Calculated using the mass fraction of the minority component and the following densities at 140![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) °C PS=0.969 g cm−3, PLA=1.154 g cm−3, PEO=1.064 g cm−3.
e Principal spacing from SAXS analysis at 20°C. °C PS=0.969 g cm−3, PLA=1.154 g cm−3, PEO=1.064 g cm−3.
e Principal spacing from SAXS analysis at 20°C.
|
|||||||
| SL1 | 21.7 | 11.1 | 32.8 | 33.7 | 1.09 | 0.300 | 27.8 |
| SL2 | 41.1 | 19.4 | 60.5 | 61.4 | 1.11 | 0.284 | 43.9 |
| SO1 | 15.5 | 14.0 | 29.5 | 30.4 | 1.08 | 0.452 | 24.6 |
| SO2 | 29.8 | 25.7 | 55.5 | 57.1 | 1.09 | 0.440 | 40.7 |
Small-angle X-ray scattering (SAXS) was used to determine the morphology of the parent materials. After annealing the samples at high temperature (120°C for 20 min and then 60°C for 20 min), SAXS data were acquired at 20°C (see the Experimental section for details). The one-dimensional data (intensity vs. scattering wave vector q) for the block copolymers is shown in Fig. 2. From the position of the scattering peaks and consideration of the PS volume fractions we have assigned both SL1 and SL2 as hexagonally packed cylinders of PLA in a PS matrix and both SO1 and SO2 as alternating lamellae of PS and PEO. These assignments are consistent with the phase behavior of these block copolymers reported in the literature.33,54 The principal domain spacing at 20°C for the parent block copolymers is also given in Table 1.
 | ||
Fig. 2 1D SAXS profiles of the parent diblock copolymers: PS-PLA and PS-PEO (see Table 1 and the Experimental section). (A)
SL1
(B)
SL2
(C)
SO1
(D)
SO2. The expected reflections for a cylindrical microstructure are marked by triangles (√3∶√4∶√7∶√9) and the expected reflections for a lamellar microstructure are marked by diamonds (√4∶√9∶ ). ). | ||
Characterization of PS-PLA/PS-PEO binary blends
Block copolymers SL1 and SL2 were solution blended with SO1 and SO2, respectively, to form a dozen optically transparent composites (see the Experimental section). The blend compositions were determined gravimetrically and agreed with 1H NMR spectroscopic analysis. In all of the blends the volume fraction of PS was between 0.66 and 0.72. Provided the PLA and PEO homogenously mixed, all blends were likely to be composed of cylinders of mixed PLA/PEO in a matrix of PS. In addition to the optical clarity, SAXS analysis of the blends revealed a pronounced principal scattering peak and often higher order reflections consistent with microphase separation in these composites (see below). A complete compositional profile is given in Table 2.| Blenda | f PS b | SOx (wt.%)c | PEO (wt.%)d |
|---|---|---|---|
|
a The block copolymers in parentheses are the parent materials used in the blend, see Table 1.
b Volume fraction of PS was calculated using the following densities at 140°C ρPS=0.969 g cm−3, ρPLA=1.154 g cm−3, and ρPEO=1.064 g cm−3
(see the Experimental section).
c Weight percent of SO1 or SO2 in the blend determined gravimetrically.
d Weight percent of polyethylene oxide in the blend.
|
|||
| 1(SL1/SO1) | 0.696 | 2.5 | 1.2 |
| 2(SL1/SO1) | 0.689 | 7.6 | 3.6 |
| 3(SL1/SO1) | 0.685 | 9.9 | 4.7 |
| 4(SL1/SO1) | 0.669 | 20.5 | 9.8 |
| 5(SL2/SO2) | 0.715 | 1.0 | 0.5 |
| 6(SL2/SO2) | 0.712 | 2.5 | 1.2 |
| 7(SL2/SO2) | 0.704 | 7.4 | 3.4 |
| 8(SL2/SO2) | 0.697 | 12.3 | 5.7 |
| 9(SL2/SO2) | 0.693 | 14.9 | 6.9 |
| 10(SL2/SO2) | 0.689 | 17.3 | 8.0 |
| 11(SL2/SO2) | 0.684 | 20.1 | 9.3 |
| 12(SL2/SO2) | 0.676 | 25.3 | 11.7 |
One-dimensional SAXS patterns for a representative set of SL1/SO1 blends are shown in Fig. 3 along with the parent material SL1. The principal spacing in these materials remained essentially constant at 28 nm in going from 0% SO1 (i.e., pure SL1) to 20.5% SO1 (blend 4). The complete set of data is given in Table 3. In each of the blends, scattering consistent with a hexagonally packed cylindrical morphology was observed. The other set of blends (SL2/SO2, blends 5–12) gave similar results, although higher order reflections were more difficult to observe. To establish the cylindrical morphology in these blends, scanning electron microscopy (SEM) images were acquired on thin film samples. A thin film of blend 11 was cast from toluene on a Si substrate and stained with RuO4 to provide domain contrast. The resultant SEM image is shown in Fig. 4. The dark worm-like features are likely the mixed PEO/PLA cylinders packed in a PS matrix. The average center-to-center distance for the cylinders was measured to be 49.2 nm from the SEM image, consistent with the corresponding value from SAXS of 51.8 nm.
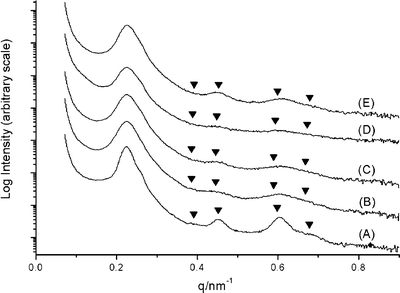 | ||
| Fig. 3 1D SAXS profiles of the SL1/SO1 blends (see Table 2 and the Experimental section). (A) SL1 (B) blend 1 (C) blend 2 (D) blend 3 (E) blend 4. The expected reflections for a cylindrical microstructure are marked by triangles (√3∶√4∶√7∶√9). | ||
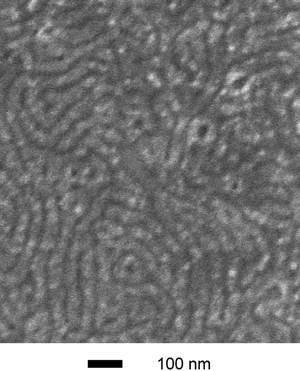 | ||
| Fig. 4 SEM image of a thin film sample. Blend 11 was spin-cast from a 15 mg ml−1 toluene solution and then stained with RuO4 for 5 min (the second largest PEO-containing blend). The SEM analysis was made on a high-resolution Hitachi S-900 FE-SEM using accelerating voltages of 1 keV. | ||
| Blenda | D pre/nmb | D post/nmc | d pore/nmd | d pore/nme |
|---|---|---|---|---|
| a The block copolymers in parentheses are the parent materials used in the blend, see Table 1. b Principal domain spacing of the blends pre-degradation by SAXS. c Principal domain spacing of the blends post-degradation by SAXS. d Diameter of nanopores in degraded blends determined from the principle spacings of the degraded blends and the volume fraction of PS and PEO in the blends (see Table 1). e Nanopore diameters (± one standard deviation) as measured in the corresponding SEM images (see the Experimental section). | ||||
| 1(SL1/SO1) | 28.1 | 27.5 | 18.1 | 13.8±0.9 |
| 2(SL1/SO1) | 28.1 | 29.8 | 19.0 | 14.6±2.2 |
| 3(SL1/SO1) | 28.0 | 27.4 | 17.3 | 14.4±1.7 |
| 4(SL1/SO1) | 27.7 | 28.4 | 16.8 | 15.3±3.0 |
| 5(SL2/SO2) | 39.7 | 41.0 | 26.4 | 18.6±4.0 |
| 6(SL2/SO2) | 44.5 | 47.2 | 30.1 | 18.3±3.8 |
| 7(SL2/SO2) | 41.6 | 41.9 | 26.0 | 19.9±5.1 |
| 8(SL2/SO2) | 40.0 | 42.7 | 25.9 | 19.4±3.8 |
| 9(SL2/SO2) | 43.0 | 43.0 | 25.6 | 18.4±3.7 |
| 10(SL2/SO2) | 42.1 | 45.2 | 26.5 | 19.5±4.7 |
| 11(SL2/SO2) | 44.9 | 49.4 | 28.5 | 19.0±4.2 |
| 12(SL2/SO2) | 40.8 | 45.8 | 25.6 | 19.7±4.4 |
To further understand the solid-state morphology of the blends, we obtained the wide-angle X-ray scattering (WAXS) and differential scanning calorimetry data (DSC) on the samples. While the WAXS data for SO2 showed clear evidence of PEO crystallinity,54,62 no PEO crystallinity was observed in the following blends: 1, 3, and 12 (samples tested). Even in the blend containing the highest fraction of PEO (12), no sharp peaks were observed by WAXS compared to the parent SO2 (Fig. 5). The lack of a WAXS signature for PEO crystalline domains in the blends is consistent with suppression of crystallinity due to the miscibility of PEO and PLA.43–53
 | ||
| Fig. 5 WAXS data for (A)
SO2 and (B) blend 12. Experiments were performed on a Siemens D5005 X-ray diffractometer with Cu Kα radiation (λ=1.542 Å) and a 0.05°
(2θ) scanning step. | ||
The lack of any significant melting endotherms in the DSC analysis of the blends 1, 3, 11, and 12 also supports the absence of PEO crystallinity, again consistent with complete mixing of the PEO and PLA in the microdomains of these composites. A representative DSC trace for a SL2/SO2 blend (blend 11) is shown in Fig. 6. The glass transition temperatures (Tgs) for PEO and PS in the parent SO2 were found to be −43 and 100°C, respectively. Likewise, the Tgs for PLA and PS in the parent SL2 were 52 and 105°C, respectively. In blend 11, two Tgs were observed at approximately 12 and 100°C. These transitions are attributed to the Tg of the miscible PEO/PLA phase and the PS phase (containing PS from both the SL and the SO block copolymers), respectively. Using the weight fractions of PEO and PLA in blend 11, the two parent Tgs, and the Fox equation for a miscible blend63 we calculated a Tg of 20°C for the mixed PEO/PLA phase, in rough agreement with the observed transition. All of the characterization data on the SL/SO blends strongly suggest that the samples consist of mixed and amorphous PLA/PEO cylinders embedded in an amorphous PS matrix.
 | ||
| Fig. 6 DSC trace of blend 11
(10°C min−1, heating). The Tg for the mixed polyethylene oxide/polylactide phase is centered at 12°C and the Tg for the polystyrene block is centered at 100°C. | ||
Solid-state processing of the PS-PLA/PS-PEO blends
All of the blends were pressed in a channel die at 140°C to orient the cylinders in the flow direction. This processing procedure was employed for PS-PLA materials and generally gave a high degree of cylinder orientation.33 Two-dimensional SAXS data of the blends after channel die processing showed two-spot patterns consistent with cylinder orientation in the flow direction. The degree of alignment in all of the blends was characterized by the second order orientation factor F2.64 The F2 values were generally greater than 0.70 indicating a high degree of orientation. Small pieces of the oriented blends were subjected to a water/methanol sodium hydroxide solution to degrade the PLA for about one week at 65°C. These monoliths were then washed with methanol and DI water, and dried overnight under reduced pressure at room temperature to afford polyether functionalized nanoporous polystyrene solids.
Characterization of the degraded monoliths
The degraded monoliths could be readily dissolved in chloroform and analyzed by 1H NMR spectroscopy. Fig. 7 shows a comparison of the 1H NMR spectra for SL1, SO1, and blend 3 both before and after degradation. There are no resonances from the PLA component (methine protons at 5.2 ppm) in the degraded blend indicating complete removal of the PLA from the monolith. Also, the degraded blend contains a distinct PEO component (methylene protons at 3.6 ppm). Integration of the PS resonances (between 6.3 and 7.2 ppm) and the PEO resonances, gave PEO compositions in the degraded blends that were generally consistent with the original blend composition data in Table 2.65 Furthermore, IR spectroscopic analysis of the degraded monoliths confirmed both the absence of PLA and the presence of PEO in the degraded blends again corroborating the complete removal of only PLA from the monoliths.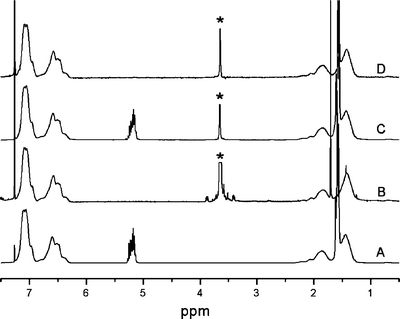 | ||
| Fig. 7 1H NMR spectra for (A) SL1, (B) SO1, (C) blend 3, and (D) degraded blend 3. * PEO resonances were truncated for clarity. | ||
SAXS analysis of the degraded blends showed a similar two spot pattern (in the shear gradient direction) as in the parent blends. A representative comparison of the one-dimensional and two-dimensional SAXS data between SL1, an SL1/SO1 blend (blend 3), and the corresponding degraded sample is shown in Fig. 8. The degraded sample and the parent blend have essentially the same principal spacing. While the higher order reflections are certainly less distinct in both the parent blend and the degraded sample compared to the parent SL1, the SAXS data support no significant change in ordered state symmetry, spacing, or microstructural alignment. In all of the degraded samples, the intensity of the principal scattering peak was significantly greater than the intensity pre-degradation, consistent with increased scattering contrast (see ref. 33).
 | ||
| Fig. 8 1D SAXS profiles for (A) SL1, (B) blend 3, and (C) degraded blend 3. The expected reflections for a cylindrical microstructure are marked by triangles (√3∶√4∶√7∶√9). The corresponding 2D SAXS patterns are also shown from the shear gradient direction (see the Experimental section). | ||
The degraded monolithic samples were further characterized by SEM. Two representative images are shown in Fig. 9 (blends 3 and 11). These images confirmed that the degraded monoliths consist of hexagonally-packed nanoscopic channels with a relatively narrow pore size distribution. The average pore sizes were measured using digital image analysis software, giving values in general agreement with those measured by SAXS experiments considering the estimated 2–3 nm coating of Pt on these samples (see the Experimental section). However, inspection clearly reveals defects present in the SEM images. This is reflected in the significant standard deviations in the average values given in Table 3.
 | ||
| Fig. 9 SEM images of degraded blends (see the Experimental section). (a) A nanoporous monolith made from blend 3, (b) A nanoporous monolith made from blend 11. | ||
DSC measurements were performed on selected degraded samples to confirm the presence of PEO in the nanoporous monoliths. Generally, samples with low PEO contents did not show any melting transitions, suggesting an amorphous PEO coating on the pore walls. In contrast, analysis of a degraded sample with high PEO loading (blend 12) gave an endothermic peak centered around 50°C. Upon integration of that peak a level of PEO crystallinity of approximately 14% was estimated.39,66 Although the DSC data are consistent with PEO crystallinity in this degraded sample, no obvious diffraction peaks were identified by WAXS results, possibly due to the relatively small fraction of PEO in the composite. Taken together, the characterization data strongly support the successful preparation of ordered nanoporous PS monoliths with PEO-coated pore walls.
Evaluation of pore-wall hydrophilicity
Amaranth, a red water-soluble dye, was used to demonstrate the significantly enhanced water compatibility of the PEO-coated nanoporous polystyrene. Standard degradation of the parent SL2 gave a nanoporous material with no PEO incorporation. This material and a nanoporous monolith from the degradation of blend 12 were placed into an aqueous solution of amaranth (1.4 mg mL−1). The nanoporous material from SL2 (containing no PEO) floated on the surface of the water and imbibed none of the aqueous solution.67 In contrast, the dye solution filled the voids in the degraded sample of blend 12 (containing less than 12 wt.% PEO) and the increased density of the material rendered it no longer buoyant.68 After 5 days,69 samples were taken out of the dye solution, washed rigorously with DI water, and then dried in a vacuum oven at room temperature overnight. The whole monolith and fractured samples containing PEO were dark red and the parent material was essentially colorless.The water compatibility of the PEO lined nanoporous materials was also investigated as a function of PEO content in three degraded blends with essentially the same pore size (blends 1, 3, and 4), through simple buoyancy experiments. The three PEO containing nanoporous materials and the parent nanoporous material from SL1 were placed in four separate vials containing pure water. Consistent with all other experiments using non-PEO coated nanoporous materials, the nanoporous material derived from SL1 floated on the surface of the water indefinitely. The degraded blends all sank to the bottom of the vessel after different times: 1 (62 h), 3 (14 h), and 4 (2.3 h). These times are correlated to the PEO content in each of the nanoporous materials; the time to sink (i.e., when the density of the water-filled monolith is greater than 1.0 g·cm−3) is inversely proportional to the PEO weight percent in these three blends. This phenomenon is currently under investigation.
Conclusions
We report a simple methodology for the preparation of ordered nanoporous PS using AB/AC diblock copolymer blends. The key components are a high Tg continuous matrix material (PS), a readily degradable minority component (PLA), and a material that is miscible with the sacrificial block, non-degradable, and water-compatible (PEO). We demonstrated that PS-PLA and PS-PEO meet these requirements, and corresponding binary blends of these two materials can be readily degraded to give a series of nanoporous materials with remarkable enhancements in water compatibility. The nanoporous plastics described in this manuscript will significantly expand applicability of ordered nanoporous materials derived from block copolymer precursors.Experimental
Materials
All reagents and solvents were used as received unless otherwise noted. Two PS-PEO (SO1, SO2) and two PS-PLA (SL1, SL2) block copolymers were synthesized through two-step sequential polymerization methods previously reported.33,60 These polymerization schemes employ the re-initiation of hydroxyl-terminated polystyrene for the subsequent growth of PEO (anionically via the potassium alkoxide) or PLA (“coordination–insertion” via triethyl aluminum catalyst) to form the final diblock copolymer.Blend preparation
PS-PEO and PS-PLA blends were prepared through co-dissolution of the block copolymer samples in CH2Cl2 at room temperature, followed by evaporation of solvent at atmospheric pressure in air. Samples were subsequently dried in vacuo at 50°C to ensure complete removal of solvent. All resulting films were colorless and transparent. Literature values for densities were used for PS,70 PEO,70 and PLA.71
General characterization methods
Size exclusion chromatography (SEC) data were collected using a Hewlett-Packard 1100 series liquid chromatograph equipped with a Hewlett-Packard 1047A refractive index detector and three Jordi polydivinylbenzene columns (pore sizes of 104, 103, and 500 Å). Samples were run at 40°C in HPLC grade THF at 1 mL min−1. Polydispersity calculations were based on instrument calibration with polystyrene standards (Polymer Laboratories). 1H NMR spectra were collected on a Varian Inova VI-500 spectrometer in CDCl3
(Cambridge) at room temperature. Differential scanning calorimetry (DSC) measurements were performed on a Q1000 Autosampler (Thermal Analysis) calibrated with an Indium standard. Parent block copolymers and blends were equilibrated at 150°C, above all thermal transitions. The degraded nanoporous monoliths were equilibrated at −100°C, to maintain the integrity of the structure prior to the thermal scan. Samples were run at 10°C min−1 under a nitrogen purge. IR spectra were acquired on a Nicolet Magna-IR spectrometer 550. Samples were crushed with dried KBr (1∶10 by volume) using a mortar and pestle and then pressed into pellets with a screw-press.
X-ray scattering
Small-angle X-ray scattering (SAXS) measurements were performed at the University of Minnesota 2.3 m and 3.5 m SAXS lines. Cu Kα X-rays (λ=1.542 Å) were generated by a Rigaku RU-200BVH rotating anode equipped with a 0.2×2 mm microfocus cathode and Franks mirror optics. Two-dimensional scattering data were recorded on a Siemens multi-wire area detector and corrected for detector response characteristics prior to analysis. Before degradation, samples were annealed 120°C (20 min), 60°C (20 min), and room temperature (10 min)
in situ under vacuum, before a 10 min exposure at room temperature. The same procedure was used on the degraded monoliths, except they were not heated above 60°C. Two-dimensional scattering images were azimuthally integrated to a one-dimensional plot of intensity versus scattering wavevector, q, where q=(4πλ−1)sin(θ/2), and λ and θ are the radiation wavelength and scattering angle, respectively. Second-order orientation factors (F2) were used to evaluate the long-range order in flow-field aligned samples.64
(F2=1 for perfectly aligned samples, F2=0 for samples with no long-range order.) Wide-angle X-ray scattering (WAXS) was performed on a Siemens D5005 X-ray diffractometer with Cu Kα radiation (λ=1.542 Å). Diffraction patterns were collected over 15–45°
(2θ) at a scanning increment of 0.05°
(2θ).
1H NMR and IR spectroscopic characterization
The following 1H NMR spectroscopy resonances are representative of polymeric samples reported in this study. All resonances are reported in ppm (δ) relative to CHCl3 (s, 7.27 ppm), and are either broad (b) or contain multiple overlapping peaks (m). PS-PLA: 6.2–7.4 (m, aromatic protons), 5.2 (m, –CO–CH(CH3)–O–), 1.7 (m, –CH(C6H5)–CH2–), 1.6 (m, –CO–CH(CH3)–O–). PS-PEO: 6.2–7.4 (m, aromatic protons), 3.6 (b, –O–CH2–CH2–O–), 1.7 (m, –CH(C6H5)–CH2–). In the IR spectra the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretch in PLA was observed near 1757 cm−1. C–O and C–C stretching and CH2 rocking modes present in PEO were observed near 1105 cm−1.72
O stretch in PLA was observed near 1757 cm−1. C–O and C–C stretching and CH2 rocking modes present in PEO were observed near 1105 cm−1.72
Microstructural alignment
Blends were aligned using a channel die (2 mm in width and 55 mm in length) as previously described for pure PS-PLA block copolymer samples.33 Blends were pre-pressed into a stainless steel mold lined with two Teflon sheets at 140°C. The resulting piece was placed vertically in the center of the channel die and pressed again at 140°C. The PLA/PEO composite cylinders were aligned in the direction of polymer flow as confirmed by SAXS measurements and SEM observations.
Degradation of PLA in blend monoliths
Blend monoliths formed in the channel die were placed into a sodium hydroxide water/methanol (60∶40 by volume) solution at 65°C for about one week, the temperature deliberately held just above the Tg of PLA. The monoliths were then washed with methanol and DI water, and later dried under reduced pressure at room temperature overnight.
Scanning electron microscopy (SEM)
Visualization of the nanoporous structure in the nanoporous samples was achieved through SEM of samples both perpendicular and parallel to the original channel die flow field direction. Samples were cut with a razor blade at room temperature, mounted onto brass shims with colloidal graphite (Ted Pella), and sputter-coated with 2–3 nm of platinum (the thickness of platinum was estimated from a calculated deposition rate and experimental deposition time). Thin film samples of the undegraded blends were spin-cast from a solution (15–30 mg ml−1 toluene) onto a Si substrate at 2000 rpm using a Headway Research Inc. (Model 1-EC101D-R485) photo-resist spinner. Contrast in blends was achieved through selective PS staining with RuO4 vapor (5 min exposure, 0.5% aqueous, Ted Pella) prior to SEM observation. All thin film samples were mounted onto brass shims with carbon tape and observed directly without metal coating. The SEM performed on a high-resolution Hitachi S-900 FE-SEM using accelerating voltages of 1 keV and 3 keV for thin film and degraded blends, respectively.Acknowledgements
We thank the National Science Foundation (DMR-0094144) and the David and Lucile Packard Foundation for their support of this work. The work was also supported in part by the MRSEC program of the National Science Foundation (DMR-0212302). Dr. Joon-Hyung Lee is acknowledged for providing the two PS-PEO samples. We thank Roberto Olayo-Valles for helpful discussions and technical assistance.References
- A. Adedeji, T. Grünfelder, F. S. Bates and C. W. Macosko, Polym. Eng. Sci., 1996, 36, 1707 Search PubMed.
- K. Naito, H. Hieda, M. Sakurai, Y. Kamata and K. Asakawa, IEEE Trans. Magn., 2002, 38, 1949 CrossRef CAS.
- I. W. Hamley, Angew. Chem., Int. Ed. Engl., 2003, 42, 1692 CrossRef CAS.
- M. Lazzari and M. A. López-Quintela, Adv. Mater., 2003, 15, 1583 CrossRef CAS.
- I. W. Hamley, Nanotechnology, 2003, 14, R39 CrossRef CAS.
- C. Park, J. Yoon and E. L. Thomas, Polymer, 2003, 44, 6725 CrossRef CAS.
- M. R. Buchmeiser, Angew. Chem., Int. Ed. Engl., 2001, 40, 3795 CrossRef CAS.
- T. Thurn-Albrecht, J. Schotter, G. A. Kästle, N. Emley, T. Shibauchi, L. Krusin-Elbaum, K. Guarini, C. T. Black, M. T. Tuominen and T. P. Russell, Science, 2000, 290, 2126 CrossRef CAS.
- T. Thurn-Albrecht, R. Steiner, J. DeRouchey, C. M. Stafford, E. Huang, M. Bal, M. Tuominen, C. J. Hawker and T. P. Russell, Adv. Mater., 2000, 12, 787 CrossRef CAS.
- G. Liu, J. Ding, T. Hashimoto, K. Kimishima, F. M. Winnik and S. Nigam, Chem. Mater., 1999, 11, 2233 CrossRef CAS.
- For an interesting related system see Y. Xu, W. Gu and D. L. Gin, J. Am. Chem. Soc., 2004, 126, 1616 Search PubMed.
- J.-M. Ha, J. H. Wolf, M. A. Hillmyer and M. D. Ward, J. Am. Chem. Soc., 2004, 126, 3382 CrossRef CAS.
- J.-S. Lee, A. Hirao and S. Nakahama, Macromolecules, 1988, 21, 274 CrossRef CAS.
- P. Mansky, C. K. Harrison, P. M. Chaikin, R. A. Register and N. Yao, Appl. Phys. Lett., 1996, 68, 2586 CrossRef CAS.
- T. L. Morkved, M. Lu, A. M. Urbas, E. E. Ehrichs, H. M. Jaeger, P. Mansky and T. P. Russell, Science, 1996, 273, 931 CrossRef CAS.
- T. Hashimoto, K. Tsutsumi and Y. Funaki, Langmuir, 1997, 13, 6869 CrossRef CAS.
- V. Z.-H. Chan, J. Hoffman, V. Y. Lee, H. Latrou, A. Avgeropoulos, N. Hadjichristidis, R. D. Miller and E. L. Thomas, Science, 1999, 286, 1716 CrossRef CAS.
- R. Mäki-Ontto, K. de Moel, W. de Odorico, J. Ruokolainen, M. Stamm, G. ten Brinke and O. Ikkala, Adv. Mater., 2001, 13, 117 CrossRef CAS.
- K. W. Guarini, C. T. Black and S. H. I. Yeung, Adv. Mater., 2002, 14, 1290 CrossRef CAS.
- O. Ikkala and G. ten Brinke, Science, 2002, 295, 2407 CrossRef CAS.
- K. Shin, K. A. Leach, J. T. Goldbach, D. H. Kim, J. Y. Jho, M. Tuominen, C. J. Hawker and T. P. Russell, Nano Lett., 2002, 2, 933 CrossRef CAS.
- A. M. Urbas, M. Maldovan, P. DeRege and E. L. Thomas, Adv. Mater., 2002, 14, 1850 CrossRef CAS.
- U. Jeong, D. Y. Ryu, J. K. Kim, D. H. Kim, T. P. Russell and C. J. Hawker, Adv. Mater., 2003, 15, 1247 CrossRef CAS.
- A. Sidorenko, I. Tokarev, S. Minko and M. Stamm, J. Am. Chem. Soc., 2003, 125, 12211 CrossRef CAS.
- S. H. Kim, M. J. Misner, T. Xu, M. Kimura and T. P. Russell, Adv. Mater., 2004, 16, 226 CrossRef CAS.
- K. A. Cavicchi, A. S. Zalusky, M. A. Hillmyer and T. P. Lodge, Macromol. Rapid Commun., 2004, 25, 704 CrossRef CAS.
- S. B. Lee, D. T. Mitchell, L. Trofin, T. K. Nevanen, H. Söderlund and C. R. Martin, Science, 2002, 296, 2198 CrossRef CAS.
- For a relevant example employing block copolymers see ref. 22.
- J. Oh, Y. Tak and J. Lee, Electrochem. Solid-State Lett., 2004, 7, C27 CrossRef CAS.
- T. Ohgai, L. Gravier, X. Hoffer, M. Lindeberg, K. Hjort, R. Spohr and J.-P. Ansermet, J. Phys. D: Appl. Phys., 2003, 36, 3109 CrossRef CAS.
- K. Moller and T. Bein, Chem. Mater., 1998, 10, 2950 CrossRef CAS.
- I. W. Hamley, in The Physics of Block Copolymers, Oxford University Press, 1998, ch. 2 Search PubMed.
- A. S. Zalusky, R. Olayo-Valles, J. H. Wolf and M. A. Hillmyer, J. Am. Chem. Soc., 2002, 124, 12761 CrossRef CAS.
- A. S. Zalusky, R. Olayo-Valles, C. J. Taylor and M. A. Hillmyer, J. Am. Chem. Soc., 2001, 123, 1519 CrossRef CAS.
- J. H. Wolf and M. A. Hillmyer, Langmuir, 2003, 19, 6553 CrossRef CAS.
- M. W. Matsen, Macromolecules, 1995, 28, 5765 CrossRef CAS.
- K. I. Winey, E. L. Thomas and L. J. Fetters, Macromolecules, 1991, 24, 6182 CrossRef CAS.
- T. S. Bailey, H. D. Pham and F. S. Bates, Macromolecules, 2001, 34, 6994 CrossRef CAS.
- T. S. Bailey, C. M. Hardy, T. H. Epps III and F. S. Bates, Macromolecules, 2002, 35, 7007 CrossRef CAS.
- T. H. Epps III, E. W. Cochran, C. M. Hardy, T. S. Bailey, R. S. Waletzko and F. S. Bates, Macromolecules, accepted Search PubMed.
- A. S. Zalusky, B. J. S. Johnson, S. Cho and M. A. Hillmyer, unpublished results.
- G. Liu, J. Ding and S. Stewart, Angew. Chem., Int. Ed. Engl., 1999, 38, 835 CrossRef CAS.
- H. Younes and D. Cohn, Eur. Polym. J., 1988, 24, 765 CrossRef CAS.
- T. G. Park, S. Cohen and R. Langer, Macromolecules, 1992, 25, 116 CrossRef CAS.
- A. J. Nijenhuis, E. Colstee, D. W. Grijpma and A. J. Pennings, Polymer, 1996, 37, 5849 CrossRef CAS.
- M. Sheth, R. A. Kumar, V. Davé, R. A. Gross and S. P. Mccarthy, J. Appl. Polym. Sci., 1997, 66, 1495 CrossRef CAS.
- M. Baiardo, G. Frisoni, M. Scandola, M. Rimelen, D. Lips, K. Ruffieux and E. Wintermantel, J. Appl. Polym. Sci., 2003, 90, 1731 CrossRef CAS.
- Y. Hu, M. Rogunova, V. Topolkaraev, A. Hiltner and E. Baer, Polymer, 2003, 44, 5701 CrossRef CAS.
- Y. Hu, Y. S. Hu, V. Topolkaraev, A. Hiltner and E. Baer, Polymer, 2003, 44, 5711 CrossRef CAS.
- D. Kubies, F. Rypáček, J. Kovářová and F. Lednický, Biomaterials, 2000, 21, 529 CrossRef CAS.
- W. Jiang and S. P. Schwendeman, Pharm. Res., 2001, 18, 878 CrossRef CAS.
- É. Kiss, I. Bertóti and E. I. Vargha-Butler, J. Colloid Interface Sci., 2002, 245, 91 CrossRef CAS.
- Y.-H. Na, Y. He, X. Shuai, Y. Kikkawa, Y. Doi and Y. Inoue, Biomacromolecules, 2002, 3, 1179 CrossRef CAS.
- P. Huang, L. Zhu, S. Z. D. Cheng, Q. Ge, R. P. Quirk, E. L. Thomas, B. Lotz, B. S. Hsiao, L. Liu and F. Yeh, Macromolecules, 2001, 34, 6649 CrossRef CAS.
- H. G. Jeon, S. D. Hudson, H. Ishida and S. D. Smith, Macromolecules, 1999, 32, 1803 CrossRef CAS.
- K. Kimishima, H. Jinnai and T. Hashimoto, Macromolecules, 1999, 32, 2585 CrossRef CAS.
- H. Frielinghaus, N. Hermsdorf, K. Almdal, K. Mortensen, L. Messé, L. Corvazier, J. P. A. Fairclough, A. J. Ryan, P. D. Olmsted and I. W. Hamley, Europhys. Lett., 2001, 53, 680 CrossRef CAS.
- H. Frielinghaus, N. Hermsdorf, R. Sigel, K. Almdal, K. Mortensen, I. W. Hamley, L. Messé, L. Corvazier, A. J. Ryan, D. van Dusschoten, M. Wilhelm, G. Floudas and G. Fytas, Macromolecules, 2001, 34, 4907 CrossRef CAS.
- N. Y. Vaidya and C. D. Han, Macromolecules, 2000, 33, 3009 CrossRef CAS.
- R. P. Quirk and J. J. Ma, J. Polym. Sci. Part A: Polym. Chem., 1988, 26, 2031 CrossRef CAS.
- H. Frielinghaus, W. B. Pedersen, P. S. Larsen, K. Almdal and K. Mortensen, Macromolecules, 2001, 34, 1096 CrossRef CAS.
- L. Zhu, S. Z. D. Cheng, B. H. Calhoun, Q. Ge, R. P. Quirk, E. L. Thomas, B. S. Hsiao, F. Yeh and B. Lotz, J. Am. Chem. Soc., 2000, 122, 5957 CrossRef CAS.
- T. G. Fox, Bull. Am. Phys. Soc., 1956, 1, 123 Search PubMed.
- P. G. deGennes and J. Prost, The Physics of Liquid Crystals, Oxford University Press, New York, 1993 Search PubMed.
- A few of the SL2/SO2 series blends showed some discrepancy between the PEO content in the original material and the degraded material. This is possibly due to inadvertent overheating during channel die alignment resulting in processing temperatures above the “normal” processing temperature of 140°C described in the Experimental section. In separate experiments we determined that there was some minor decomposition (i.e., chain cleavage) of the PEO in these blends after this errant heating. Furthermore, we did identify some PEO homopolymer contamination in the parent SO2 material and this would be present in the blend as prepared, but not in the degraded blends. We were able to reproduce the data on these blends using the same materials, however we have since prepared new SL and SO materials for future experiments.
- ΔHf=203 J g−1, see: B. Wunderlich, Macromolecular Physics, Academic Press, New York, 1980, vol. 3, p. 67 Search PubMed.
- The estimated density of this nanoporous PS is approximately 0.6 g cm−3 (see ref. 32).
- The density of pure PS at room temperature is about 1.05 g cm−3.
- The degraded sample of SL2 remained buoyant for more than three months.
- L. J. Fetters, D. J. Lohse, D. Richter, T. A. Witten and A. Zirkel, Macromolecules, 1994, 27, 4639 CrossRef CAS.
- D. R. Witzke, R. Narayan and J. J. Kolstad, Macromolecules, 1997, 30, 7075 CrossRef CAS.
- F. E. Bailey Jr. and J. V. Koleske, Poly(ethylene oxide), Academic Press, Inc., New York, 1976, p. 115 Search PubMed.
Footnote |
| † Present address: Department of Chemistry, The College of St. Scholastica, Duluth, Minnesota, USA. |
| This journal is © The Royal Society of Chemistry 2005 |