Increasing atmospheric antimony contamination in the northern hemisphere: snow and ice evidence from Devon Island, Arctic Canada†
Michael
Krachler
*a,
James
Zheng
ab,
Roy
Koerner
b,
Christian
Zdanowicz
b,
David
Fisher
b and
William
Shotyk
a
aInstitute of Environmental Geochemistry, University of Heidelberg, Im Neuenheimer Feld 236, 69120, Heidelberg, Germany. E-mail: krachler@ugc.uni-heidelberg.de; Fax: +49 62 21 54 52 28; Tel: +49 62 21 54 48 48 Web: http://www.uni-heidelberg.de/institute/fak12/ugc/mkrachler/krachler.htm
bGSC Northern Canada, Geological Survey of Canada, Ottawa, Canada
First published on 19th October 2005
Abstract
Adopting recently developed clean laboratory techniques, antimony (Sb) and scandium (Sc) deposition were measured in a 63.72 m-long ice core (1842–1996) and a 5 m deep snow pit (1994–2004) collected on Devon Island, Canadian High Arctic. Antimony concentrations ranged from 0.07 to 108 pg g−1 with a median of 0.98 pg g−1 (N = 510). Scandium, used as a conservative reference element, revealed that dust inputs were effectively constant during the last 160 years. The atmospheric Sb signal preserved in the ice core reflects contamination from industrialisation, the economic boom which followed WWII, as well as the comparatively recent introduction of flue gas filter technologies and emission reduction efforts. Natural contributions to the total Sb inventory are negligible, meaning that anthropogenic emissions have dominated atmospheric Sb deposition throughout the entire period. The seasonal resolution of the snow pit showed that aerosols deposited during the Arctic winter, when air masses are derived mainly from Eurasia, show the greatest Sb concentrations. Deposition during summer, when air masses come mainly from North America, is still enriched in Sb, but less so. Snow and ice provide unambiguous evidence that enrichments of Sb in Arctic air have increased 50% during the past three decades, with two-thirds being deposited during winter. Most Sb is produced in Asia, primarily from Sb sulfides such as stibnite (Sb2S3), but also as a by-product of lead and copper smelting. In addition there is a growing worldwide use of Sb in automobile brake pads, plastics and flame retardants. In contrast to Pb which has gone into decline during the same interval because of the gradual elimination of gasoline lead additives, the enrichments of Sb have been increasing and today clearly exceed those of Pb. Given that the toxicity of Sb is comparable to that of Pb, Sb has now replaced Pb in the rank of potentially toxic trace metals in the Arctic atmosphere.
 James Zheng James Zheng | Michael Krachler was born in Austria, in 1967. He received both his M.Sc. in chemistry in 1993 and a PhD in analytical chemistry from the University of Graz, Austria in 1997. In 1998 he joined the Research Centre of Juelich, Germany as a Marie Curie Fellow funded by the European Commission. Since 2000 he has been a Research Associate at the University of Heidelberg and is responsible for the new trace element and ICP-MS laboratories. His current research interests centre on applications of ICP-sector field-MS for the determination of trace elements and lead isotopes in various environmental compartments. |
Aim of the investigation
Metals are ubiquitous in our modern world, and toxic metals such as Cd and Pb ranking high among major pollutants worldwide. In order to mitigate the ecological and toxicological impact of the metals, better knowledge is required of their sources, pathways, mobility and fate in the surficial environment. A challenge that commonly faces environmental researchers is to distinguish between lithogenic and anthropogenic contributions of metal accumulation. While industrial emissions of many trace elements have declined in recent decades (e.g. Pb), they have done so from levels which were extremely high, relative to natural, background deposition rates. There are still major gaps in our knowledge of natural sources of atmospheric trace metals, and their variability at different time scales. In that context, ice cores are commonly exploited as valuable environmental archives of atmospheric metal deposition.1–10Because antimony (Sb) is present in the Earth’s crust at concentrations below 1 mg kg−1,11 there have been few quantitative studies of the geochemical cycle of this potentially toxic element.12 Recognizing that Sb2O3 is a suspected carcinogen, the European Commission and the US Environmental Protection Agency have listed Sb and its compounds as “priority pollutants”.13,14 With the progressive removal of asbestos from brake pads in the 1980’s and its partial replacement by Sb2S3, today Sb is the single most highly enriched element in urban dusts.15 Studies have shown that Sb in plants growing beside motorways and in urban areas is almost exclusively related to vehicular traffic and is found mainly in the thoracic particle fraction (i.e., 10–2.5 μm) and in the respirable fraction (i.e., <2.5 μm).16–18 These fine dust particles, released to the atmosphere through abrasion from Sb containing brake pads, show the greatest Sb enrichments.16
Worldwide, two-thirds of total Sb production is used in the manufacture of flame retardants (as Sb2O3,) for plastics and textiles, most of which eventually ends up in a non-recyclable waste stream. In addition, Sb2O3 is used in the catalysis of PET, and the incineration of this material and other Sb-containing plastics (e.g. PVC) yields aerosols with an average particle diameter below 1 μm.19
Measurements of Sb in peat cores from rain-fed bogs in Switzerland, Scotland, and the Shetland Islands have shown that the chronology and intensity of atmospheric Sb contamination in Europe since the Roman period is similar to that of Pb.15 In other words, Sb data from peat core analyses reveal that (i) atmospheric Sb contamination has a history of at least 2000 years and (ii) Sb was subjected to long-range transport which extended contamination even to remote regions of Europe. However, the extent of Sb contamination remains unclear and the possible importance of Sb as a global pollutant is not yet established. While many of the trace metals of contemporary environmental interest show declining concentrations and enrichments in recent layers of polar ice,1,5 there is very little Sb data available for comparison.8 To elucidate the potential relevance of Sb as a global pollutant, investigations of atmospheric Sb in remote locations such as the Arctic are needed.
Adopting clean laboratory procedures, which we have developed and continue to improve,20–22 a total of 510 age-dated ice and snow samples from Devon Island, Arctic Canada were analysed for Sb, scandium (Sc) and Pb using inductively coupled plasma sector field mass spectrometry (ICP-SMS). The detection limits which we have obtained for Sb (30 fg g−1) and Sc (5 fg g−1) have allowed these elements to be measured in polar ice for the first time. Using Sc as a conservative, lithogenic tracer of natural mineral dusts, we employ the Sb/Sc ratio of crustal rocks, soils, and pre-anthropogenic aerosols from a peat bog (6000 to 9000 years old) to try to determine the extent to which Sb is enriched in the Arctic atmosphere, and how these enrichments have evolved during the past ca. 160 years. Our main objective is to test the hypothesis presented earlier that Sb, like Pb, is a global atmospheric contaminant.15 In addition, using 45 samples from a pit which has accumulated 5 m of snow during the past ten years (1994 to 2004), we are able to examine and illustrate the pronounced seasonal variations in atmospheric Sb deposition. Meteorological data is then used to attribute the modern inputs to predominant source areas. Using available production statistics, the most likely sources of atmospheric Sb contamination are identified.
Experimental
Laboratories and instrumentation
To minimize the potential risk of contamination, all sample handling and the preparation of all standards were performed in clean rooms under laminar flow clean air benches of at least class 100. Decontamination of the ice samples was carried out in a certified cold clean room of class 100. The ice and snow samples were melted, acidified with high purity HNO3 (68–72%) to 0.5% (v/v) and bottled in a class 10 clean air cabinet at the Geological Survey of Canada (GSC) or at the University of Heidelberg.All ICP-MS measurements were carried out with an Element2 ICP-SMS (Thermo Electron, Bremen, Germany) operated in a class 1000 clean laboratory. A micro volume autosampler (ASX 100, Cetac Technologies, Omaha, NE, USA), a high efficiency sample introduction system (Apex Q, Elemental Scientific Inc., Omaha, NE, USA) and a sapphire injector tube were employed to transport the analytes into the plasma of the ICP-SMS. The autosampler and the Apex sample introduction system were hosted in a class 100 laminar flow bench. Details of all analytical procedures including ICP-SMS operating conditions and the data acquisition parameters have been reported earlier.20–22
Reagents and standards
For the preparation of all solutions, high purity water (18.2 MΩ cm) from a MilliQ-Element system (Millipore, Milford, MA, USA) designed for ultra trace analysis was used. Nitric acid (65%, analytical-reagent grade, Merck, Darmstadt, Germany) was further purified twice by distillation, using a high purity quartz unit for sub-boiling of acids. (MLS GmbH, Leutkirch, Germany). Both the water purification system and the sub-boiling distillation unit were operated in clean rooms.Calibration solutions for Sb, Sc and Pb were prepared daily by appropriate dilution of 10 mg l−1 stock standard solutions (Merck) with 0.5% (v/v) high-purity nitric acid. Quantification of trace element concentrations was performed by linear regression of the calibration curves.
Collection of ice and snow samples and sample treatment
A 63.72-m long firn core (D2000) was drilled in May 2000 from the Devon ice cap (Fig. 1, Devon Island; 75° N; 82° W; 1860 m asl). The drilling site presently experiences a mean annual surface temperature of −23 °C, an accumulation rate of 0.28 m year−1 (ice equivalent based on 40 year mass balance records) and 20% summer melt (mass). To minimize potential metal contamination of the core, the electro-mechanical auger was modified by replacing steel and aluminium components of the sonde with titanium and polyethylene parts. The modified auger was so designed that the core was directly drilled into an acid-cleaned, high-density polyethylene (HDPE) tube inserted in the drill barrel. Once a core was drilled, the tube was removed, capped and sealed in a polyethylene bag. There was no other contact with the cores except occasionally, using gold-plated tongs. Detailed information on the new clean drill made from high purity titanium can be found elsewhere.23,24 | ||
| Fig. 1 Map of a selected area of the Canadian High Arctic highlighting Devon Ice Cap where the ice core and the snow pit were collected at 75° N; 82° W; 1860 m asl in the years 2000 and 2004, respectively. | ||
To evaluate deposition trends since the core was drilled, a snow pit was dug on Devon Island in the spring of 2004. The 5 m snow pit representing precipitation from 1994 to 2004 was dug by hand using stainless steel shovels. The sampling pit wall was further cleaned using titanium chisels right before sampling. The purpose of the pit was to evaluate the recent metal pollution trends in the Arctic since the D2000 core was drilled. Personnel working in the pit wore Tyvek clean-room jackets during the digging and sampling. Samples were taken from the pit in intervals ranging from 6.4 to 19.1 cm for firn layers. The top sample (fresh snow) was taken from outside of the pit (about 1 cm thickness of snow collected at approximately 10 m in front of the pit). All samples were taken with a pre-cleaned titanium chisel and a pre-cleaned HDPE scoop. Before each sampling, the titanium chisel and scoop were further cleaned by pushing in and out of snow on the sidewall in the pit to remove any possible dusts on their surfaces. Samples were directly taken into 500 ml pre-cleaned HDPE bottles, which were kept capped all the time before and after sampling. Cleaning procedures for bottles and utensils were as described by Zheng et al.24 A total of 45 samples were taken from the pit and they were shipped frozen to the University of Heidelberg laboratories for further processing and analyses. Clean chambers used for this processing are better than class 10 and those clean chambers are located in a metal-free class 100 clean room. Right before analysis, the samples were acidified with high purity nitric acid when they were half-melted and then transferred to autosampler cups with pre-cleaned HDPE bottles. To avoid localised leaching from beaker walls during sample acidification/melting and for convenience, a 1 + 1 dilution of concentrated nitric acid with high purity water was used. Therefore, the acidification of ice samples was accomplished by adding 0.08 ml of 1 + 1 diluted high purity nitric acid per every 10 ml of ice sample, which resulted in a pH of the melted ice samples of approximately 1.2. Ice samples were decontaminated manually and processed similarly in the clean laboratories in Ottawa.24
The cleanliness of the Ti drill and effectiveness of the decontamination procedure of the ice core sections was investigated in detail demonstrating that the Ti drill “yields” ice cores that are distinctly less contaminated than those obtained by other types of conventional drills used worldwide. Additionally an inter-laboratory study revealed that leaching of trace elements from the storage containers is well under control and that procedural blanks are so low that they do not measurably affect trace element concentrations in the ice samples.24
The chronology for the D2000 core was derived from another, deeper core (300-m long) drilled in 1998 (D1998) from the same site. The depth-age relationship is based on annual layer counting using seasonal variations in δ18O and major ions (Na+, SO42− and NO3−), and on distinctive time markers such as SO42− and acidity peaks related to the Laki (Iceland; 1783 AD), Tambora (Indonesia; 1816 AD) and) Katmai (Alaska; 1912 AD) volcanic eruptions. Correlation between the 1998 and 2000 cores was made using the 1958 (16.5 m in real depth below surface) and 1963 (13.5 meters) radioactive horizons as well as the 1998 age dating model. Based on this correlation, the age of the ice at the bottom of D2000 core (63.72 m) corresponds to AD 1842. The dating error for first top 20 m is estimated to be ±1 year; ±2 years for depths between 20 and 40 m and ±5 years for the remainder of the core. Depth and age relationships for the pit were developed by examination of ice layers and the pit stratigraphy. The dating error for the pit is estimated to be less than six months.
Quality control
As no certified reference material for trace elements in polar ice is currently available, a riverine water reference material (SLRS-4, National Research Council Canada, Ottawa, Canada) with lowest available Sb, Sc and Pb concentrations was used for quality control purposes. Good agreement between the experimentally established and certified (Pb, Sb) and reported (Sc) concentrations was established.Results and discussion
Quality control and detection limits
In contrast to our previous work,20–22 a high efficiency sample introduction system (Apex Q) was utilized for trace element analyses during this study. Using the Apex, the analytes are concentrated via desolvation of the sample aerosol which is initially heated to 140 °C followed by cooling to 2 °C before entering the plasma. The increase in sensitivity with the Apex compared to our conventional sample introduction system was approximately six-fold and helped to lower the detection limit (LOD) for Sc from 0.026 pg g−1 to 0.005 pg g−1.22 Detection limits were based on the 3σ criterion from the measurement of 16 independently prepared blank solutions containing 0.5% nitric acid and high purity water. For Sb the LOD was improved from 0.07 to 0.03 pg g−1. The higher improvement of LODs for Sc clearly indicates that the LOD for Sc is limited by counting statistics whereas the LOD for Sb is mainly determined by its blank, i.e. contamination level. The LOD for Sb is lower by one order of magnitude compared to previous ice/snow studies25 and here, Sb concentrations were greater than the LOD for all samples. Acid blanks containing 0.5% HNO3 and high purity water amounted to 7.0 ± 1.6 fg g−1 and 43 ± 10 fg g−1 for Sc and Sb, respectively.The riverine reference water material SLRS-4 with certified concentrations for Sb (230 ± 40 pg g−1) and Pb (86 ± 7 pg g−1) was analyzed at regular intervals during ice analysis. The experimentally determined Sb (239 ± 18 pg g−1; N = 40) and Pb (77 ± 10 pg g−1; N = 40) concentrations agreed well with the certified values. For Sc, no adequate certified water reference material is currently available, but previously obtained values (11.3 ± 0.6 pg g−1),22 were reproduced in this study (10.3 ± 0.9 pg g−1, N = 38).
Abundance and distribution of Sb and Sc
The concentrations of Sb in the investigated ice and snow samples (N = 510) ranged from 0.07 to 108 pg g−1, with a median of 0.98 pg g−1 (Table 1). These values are similar to those determined in 68 Greenland snow samples representing the period of 1990–1995 (range: 0.21–4.3 pg g−1, median 0.72 pg g−1),26 but are at least an order of magnitude lower than Sb concentrations found in snow and ice samples in the European Alps.8| Element | Minimum | Maximum | Median |
|---|---|---|---|
| Antimony | |||
| Devon 2000 firn core (1842–1996) | 0.07 | 108 | 0.98 |
| Devon 2004 snow pit (1994–2004) | 0.13 | 3.71 | 1.03 |
| Scandium | |||
| Devon 2000 firn core (1842–1996) | 0.02 | 8.80 | 0.47 |
| Devon 2004 snow pit (1994–2004) | 0.08 | 1.64 | 0.31 |
The ratio of maximum to minimum Sb concentrations in the present study exceeds a factor of 1500. Even though the Sb concentration pattern shows remarkable variation, a detailed chronology of past atmospheric Sb could be extracted from the entire data set either by calculating a running median or running average and using intervals of 50 data points (Fig. 2A). On plotting ice core data for trace metals, the conventional approach has been to use the running average to smooth such a fluctuating data set.8 The data shown in Fig. 2A illustrates how data processing can profoundly affect the temporal trend. The Sb concentrations obtained by averaging are dominated by 21 samples out of 510 containing Sb concentrations in the range of 5–108 pg g−1. With such “outliers” in the data set containing extraordinary Sb concentrations, the running average overestimates the Sb concentration profile, relative to the smoothed concentration pattern (Fig. 2A). The increase in Sb concentrations obtained between 1860 and 1880 A.D. calculated using a running average, for example, is an artefact caused by a single ice sample containing 108 pg Sb g−1. In contrast, the running median reveals a much smaller increase in the Sb concentrations during the same period (Fig. 2A). Regardless of whether this high concentration is related to a natural event or was caused by the sampling or processing of the ice sample, clearly, the running median provides a much more representative and conservative description of the long-term trends. Here, the running median is used exclusively throughout this study.
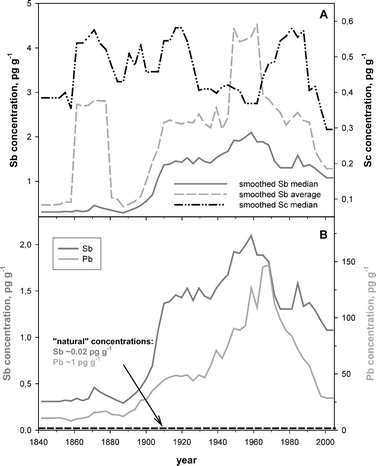 | ||
| Fig. 2 (A) Influence of data processing on the chronology of 155 years of atmospheric Sb and Sc as revealed by the Devon 2000 ice core and snow pit. Solid lines represent 50 point moving averages or medians, i.e. reflecting approximately five years of snow accumulation. (B) Chronology of antimony (Sb) and lead (Pb) in the Canadian Arctic. Data represent measurements on 510 individual ice and snow samples, i.e. ∼3 samples year−1 on average. Median concentrations of Sb and Pb together with their estimated natural concentrations are displayed. | ||
Except for 4% of the samples with Sb concentrations >5 pg g−1, the remaining ice samples fluctuated only within a range of a factor of 50 and this was mainly caused by seasonal variations (see below). It is important to note here that Sb concentrations greater than 5 pg g−1 were confirmed by independent ICP-MS measurements on the same sample aliquots at the Geological Survey of Canada in Ottawa. As contamination of the ice samples by Sb during the entire decontamination procedure is very unlikely, these high concentrations can be possibly explained by the relatively high resolution in time (on average more than three samples per year) of the ice samples. Therefore one ice sample represents only a part of the year and thus Sb concentrations may vary largely due to seasonal variations (see below). During periods of extended dry deposition (mainly in the winter season), for example, dust particles deposited onto the snow surface may be responsible for an ice sample with a distinctly elevated Sb concentration. Assuming a particle diameter of 1 μm, a particle density of 2.5 g cm−3, and a Sb concentration of 10 μg kg−1, such a single particle, in an ice volume of 10 ml, increases the actual Sb concentration by ∼0.13 pg g−1.
Interpretation of the Sb chronology
The smoothed (running median) Sb concentration profile largely follows that of Pb (Fig. 2B), with both elements showing elevated concentrations during the 20th century. In fact, the geochemical and mineralogical association of Sb with Pb minerals implies that the temporal trend of both elements should evolve more or less in parallel, largely reflecting Pb smelting.12 As chalcophile elements, both Sb and Pb are commonly enriched in coal, and because fossil fuel combustion appears to be the largest single source of anthropogenic Sb (∼50% of total Sb emissions) to the global atmosphere,27 the history and intensity of Sb and Pb emissions to the environment also are both linked to the combustion of fossil fuels in the late 19th and early 20th century.In the oldest ice samples (∼1840–1860 AD), Sb concentrations were <0.5 pg g−1 (Fig. 2B). Starting from approximately 1900 AD, Sb concentrations increased rapidly, probably reflecting the growing importance of anthropogenic activities such as coal burning, mining and smelting of Pb and Cu ores, reaching a plateau at ∼1.4 pg g−1 between 1910 and 1940 AD. In contrast to Sb, the relative increase of Pb concentrations was much smaller during the same period. The low melting point (550 °C) of stibnite (Sb2S3) relative to galena (PbS, 1114 °C) is a possible explanation for these differences. In other words, during the combustion of metal sulfides, whether from ores or in coal, larger amounts of Sb relative to Pb are emitted to the atmosphere. Leaded gasoline, an additional source of Pb, is recorded by the ice core since 1930 (Fig. 2B).
Antimony concentrations reached maximum values of ∼2 pg g−1 in the late 1950s reflecting the economic boom after WWII. Since then Sb concentrations in the snow declined, reaching ∼1.2 pg g−1 during the last decade. We assume that the pronounced decrease in Sb emissions is mainly related to the use of dedicated filter technologies in coal fired power plants that are still widely used today and the emission reduction technologies used by modern smelters. The decline in gasoline lead consumption starting in the 1970s allowed Pb concentrations to fall by 80%. In contrast, Sb concentrations declined only by 50% during the last two to three decades. The comparison of Sb/Sc with Pb/Sc from 1840 to 2000 shows that Sb was more enriched than Pb during ten of the past fifteen decades, including the last two (Fig. 3B). The decades characterised by Pb enrichments exceeding those of Sb are the ones corresponding to the greatest use of leaded gasoline (ca. 1945 to 1990). Moreover, when both Sb/Sc and Pb/Sc went into decline (ca. 1960), Sb/Sc declined more rapidly: this may reflect the effectiveness of filtration technologies reducing Sb and Pb emissions from point sources. While this probably had a comparable effect on both metals, the ongoing use of leaded gasoline reduced the rate of the decline in Pb/Sc.
To put the Sb concentrations obtained from the ice core into perspective, the “natural, lithogenic” Sb concentration (Sbnat) has been calculated as follows:
| [Sb]nat = [Sc]sample([Sb]/[Sc])UCC | (1) |
In contrast to the highly variable Sb data, Sc concentrations—that are used as a proxy of mineral dust input28—reveal no distinct temporal trend (Fig. 2A). Except for three periods of slightly elevated Sc inputs, mineral dust deposition was effectively constant over the entire period represented by the ice core and the snow pit (ca. 160 years). Aerosols from coal burning, but perhaps also from soil dust, erosion, deforestation, agriculture and construction, are possible sources of the increased Sc concentrations found during these three periods. It remains to elucidated, however, which were the predominant causes of the increased Sc deposition. Similar to Sb, the scatter in the Sc values (0.02–8.80 pg g−1) in the ice core can be attributed to seasonal changes (see below).
Enrichment factors for Sb
To assess the enrichment or depletion of Sb concentrations over time relative to the UCC, Sb enrichment factors (Sb EF) were calculated as| Sb EF = ([Sb]/[Sc])sample/([Sb]/[Sc])UCC | (2) |
Although the absolute values of the EF certainly depend on the “background” Sb/Sc ratio employed, the evolution of the temporal trend in the EF is identical regardless of the reference level used. The pronounced impact of normalizing Sb/Sc concentrations in the ice samples to their corresponding values in the upper or average continental crust is shown in Fig. 3A.11 Using these values, Sb EF as great as ∼110 and ∼260, respectively, are seen (Fig. 3A). Considering the natural background levels of Sb (8 ng g−1) and Sc (80 ng g−1) that have been established in natural dusts deposited in a peat profile from the Jura Mountains, Switzerland between ca. 9000 to 6000 calendar years BP, the maximum Sb EF amounts only to ∼50 (Fig. 3A).30 Another alternative would be to use the Sb/Sc ratio typical of soils, which is more representative of soil-derived aerosols than the corresponding values for crustal rocks (1/7).31 This approach yields a maximum Sb EF of ∼40 (Fig. 3A). It is important to note here, that regardless of the reference levels used, Sb is markedly enriched in all samples, but especially so in those corresponding to the 20th century.
 | ||
| Fig. 3 (A) Influence of background concentrations used for normalization on absolute values of Sb EF. See text for details. EGR = Etang de la Gruère, a peat bog in the Jura Mountains, Switzerland. (B) Medians of Sb/Sc and Pb/Sc ratios, respectively, reflecting the temporal change of the enrichment of Sb and Pb. The ellipse is included to guide the eye to the changes seen in Sb/Sc since ca. 1970, while the horizontal line displays the Sb/Sc and Pb/Sc ratios in the Upper Continental Crust (UCC). The differences in scales of the y axes are proportional to the differences (55×) in crustal abundance of the two elements (Pb 17 μg kg−1, Sb 0.31 μg kg−1). | ||
The concept for calculating elemental EF is based on the assumption that an appropriate reference level of the considered elements is well established and that the reference element used for normalisation is only derived either from one major source, i.e. weathering of rocks on the continental crust or marine aerosols, etc. The calculations made above, however, demonstrate the weakness and limitations of this concept. Background values for Sb and Sc on Devon Island have not yet been established. In general we prefer to use Sc as a conservative reference element, because in contrast to other lithogenic elements such as Al, Ti or Zr, Sc has no industrial use, shows no preference for specific mineral phases and is rather uniformly distributed among the dominant minerals of the Earth’s crust. Given the above mentioned difficulties in obtaining reliable, absolute Sb EF, it is obvious that site-specific background values, which can only be obtained by analyzing older ice from the same site, and their temporal variation are urgently needed.
For the time being, however, rather than calculate an absolute value for the enrichment, we avoid this altogether and simply present the Sb/Sc ratio to document its temporal evolution (Fig. 3B). For a comparison to Sb, the Pb/Sc ratio is also plotted in Fig. 3B against time. As the Sc deposition to the snow surface was roughly constant during the last 160 yrs, the evolution of both the Sb/Sc and Pb/Sc ratios provides a common basis to compare Sb and Pb.
The most striking feature of this plot, however, is the 50% increase in Sb enrichments during the last three decades. While modern state-of-the-art waste incineration plants have reduced atmospheric emissions of Sb, 670 tonnes Sb year−1 are still estimated to be released with stack gases to the atmosphere world-wide.32 It is worth mentioning here, that Sb emissions from waste incineration account for 19% of the total trace element emissions.32 In the European Union the emission standard for Sb is part of a sum parameter including As, Co, Cr, Cu, Mn, Ni, Pb, Sn, and V and the total of these may not exceed 0.5 mg m−3 for municipal solid waste incineration.33 Antimony together with As, Cd and Pb belongs to the so-called Class II elements that are vaporised during combustion but after condensation are found mainly in the fly ashes on particulates. We note that a significant part of these fine particles are in the sub-micrometre size class where dust control systems are less effective.33
In addition, the worldwide annual production of Sb has almost doubled from ∼70 000 ton in the late 1960’s to ∼120 000 ton in the year 2000 (J. O. Nriagu, personal communication). In the year 2003 global Sb production amounted to 142 000 ton per year, with most produced by China (88%), South Africa (4%), Russia (3%), Tajikistan (2%), and Bolivia (2%).12 With the increased use of Sb in various kinds of plastic (PVC, PET, etc.) and the increased waste incineration of plastic during the last 30 years, aerosols with an average particle diameter below 1 μm18 are continuously emitted and are then subjected to long-range transport. With an atmospheric residence time (1–2 weeks) comparable to that of Pb, long-range transport (several thousands of kilometres) of Sb from industrial and urban centres has clearly made its mark on the Arctic atmosphere (Fig. 3B). With the magnitude of Sb enrichments found in remote regions such as the Arctic today, one can easily imagine the extent of Sb pollution in regions where the corresponding point sources are located. For megacities such as Tokyo, for example, Sb EF of ∼21 000 have been determined in the <0.2 μm size fraction of airborne particulate matter continuously collected between 1995 and 2004.34
Seasonal variations of Sb and mineral dust depositions
Because of the excellent temporal resolution (45 samples representing ten years), the investigation of the snow pit makes it possible to study changing deposition rates due to seasonal variations in air mass sources, trajectories, and chemistry. Fig. 4A highlights the seasonal and annual changes of both Sb and Sc concentrations in the snow pit for the last decade. Data points in Fig. 4 marked with grey bars indicate the summer of the specific year which has been determined using major ion chemistry and snow stratigraphy. | ||
| Fig. 4 (A) Temporal trend and seasonal variations of Sb and Sc concentrations (pg g−1) as revealed by the 5 m snow pit collected on Devon Island in 2004. Grey bars indicate the summer of the respective year. (B) Influence of the reference level used for normalisation on the measured Sc data in the ice and snow samples on the absolute values of natural Sb. (C) Predominance of excess Sb during the last ten years revealing maximum anthropogenic Sb deposited in winter. | ||
Median concentrations of both Sb and Sc in the 5 m snow pit (representing deposition between ca. 1994–2004) are similar to the corresponding values in the ice core (Table 1). Antimony concentrations (N = 45) ranged from 0.13 to 3.71 pg g−1 with a median value of 1.03 pg g−1.
Numerous observations at arctic locations have shown that pollution levels during late winter and early spring can reach concentrations that are comparable to those observed at polluted mid-latitude locations.35 These so-called “Arctic haze episodes” occur predominantly between December and April with haze being comprised of particles no larger than 2 μm.36,37 Meteorological studies indicate that air flow into the Arctic during winter is predominantly from Eurasia, and rarely from North America.35 The strong transport into the Arctic from Eurasia during winter is caused by the presence of the climatologically persistent Siberian high pressure region resulting in a surge of polluted European air into the Arctic.37 For the rest of the year, particulate pollution is either absent, or present at much lower concentrations than in winter.37
A closer look at the snow pit data reveals that Sb concentrations are lowest during summer when the air masses reaching the Canadian Arctic mainly originate from North America.35–37 The lowest Sb concentration (0.1 pg g−1) was established in the snow sample from the summer of 2003. In contrast, maximum Sb values are often found in snow samples representing the winter seasons with air masses in the Canadian Arctic predominantly arriving from Europe and northern Asia (i.e. Eurasia).35–37 Calculating a Sb inventory for the last ten years reveals that ∼2/3 of the total Sb was deposited on Devon Island in the winter season and only ∼1/3 in the summer season.
Anthropogenic emissions of Sb from the emerging countries of Asia are believed to dominate global emissions27 and the snow pit data supports this interpretation. For example, Asian Sb emissions from primary Cu production (159 tonnes year−1) account for 50% of the worldwide emissions in this category.27 Similarly, Asian primary lead production emits 64% (86 tonnes year−1) and primary zinc production 41% (39 tonnes year−1) of global anthropogenic Sb in this category.27 As mentioned earlier, municipal waste incineration of various Sb-containing plastics also contributes to Sb emissions. A report on Sb emissions from waste incineration from the mid-1990s estimated European (33%) and Asian (42%) contributions to dominate worldwide Sb emissions (235 tonnes yr−1); this can at least partially explain the high Sb concentrations found in winter snow samples in the snow pit.27 Total global anthropogenic Sb emissions (1561 tonnes yr−1) are clearly dominated by contributions from Asia (44%), followed by North America (24%), Europe (17%) and South America (6.5%).27 Ignoring the Sb contributions from the southern hemisphere, the winter inventory of Sb in the snow pit (2/3 of the total) is remarkably consistent with European and Asian contributions to total global emissions estimated by Pacyna and Pacyna,27i.e. 61%.
The Sc concentration profile of the snow pit also shows greater concentrations in winter (Fig. 4A). Scandium, a proxy for mineral dust input to the snow pit, thus highlights that the winter season is dustier—possibly reflecting coal burning—than the summer seasons. Like the ice core, the snow pit data reveals no temporal trend of dust inputs.
Following eqn (1), the natural Sb concentration in the snow pit was calculated using several reference levels (Fig. 4B). The lowest natural Sb concentrations were obtained using the Sb/Sc ratio of the average continental crust while the soil Sb/Sc ratio31 yielded the greatest natural concentrations. Even though absolute values of the naturally derived, lithogenic Sb vary distinctly depending on the compartment used for normalization, the contribution of natural Sb to the total Sb inventory is always negligible (Fig. 4A and B). To unequivocally establish natural Sb concentrations, however, older, uncontaminated ice samples need to be analysed. This work will be carried out in the near future and this will also allow the calculation of absolute enrichment factors using site-specific natural background values.
The excess amount of Sb ([Sb]excess) can be easily calculated as
| [Sb]excess = [Sb]total − [Sb]nat | (3) |
To highlight the importance of the need to establish reliable natural background concentrations and their variation with time, the average Sb EF calculated using various reference levels are plotted in Fig. 5 for the snow pit. The difference between lowest and highest is ∼7.5, with the greatest Sb EF (234) obtained using the average composition of the continental crust and the smallest (31) obtained using soil as reference. Regardless of the kind of normalisation used, all calculations demonstrate that the Arctic today is profoundly contaminated with anthropogenic Sb. Less remote regions should be correspondingly more contaminated. Moreover, independent of the reference level employed to calculate the EF, Sb clearly is now more enriched in Arctic aerosols than Pb (Fig. 5).
 | ||
| Fig. 5 Average Sb EF and Pb EF for the period of 1994 to 2004 calculated using different reference levels. See text for details. | ||
Conclusions
The studies of peat bogs in Europe showed that anthropogenic activities have impacted the geochemical cycle of Sb to the same extent as that of Pb.12 The Arctic snow and ice data, however, shows that human impacts on the Sb cycle are global in extent and now exceed those of Pb. Given that Sb-bearing aerosols from combustion processes are smaller than 1 μm, primarily in the form of relatively soluble oxides, and that the toxicity of Sb is comparable to that of Pb, the recent trend in atmospheric Sb in the Arctic might have broader implications for human and ecosystem health worldwide.Acknowledgements
This work was supported by the European Commission (MIF1-CT-2005-008086) and the Forschungspool of the University of Heidelberg (Project: “Trace element analyses of ice cores for reconstruction of paleoclimate and human impact”). Additional financial and logistic support was received from Terrain Science Division and Metal in the Environment Program, Geological Survey of Canada. J. Z. thanks Dr. Susan Pullan for her advice and support.References
- C. F. Boutron, U. Gorlach, J.-P. Candelone, M. A. Bolshov and R. J. Delmas, Nature, 1991, 353, 153 CrossRef CAS.
- C. Barbante, A. Veysseyre, C. Ferrari, K. Van de Velde, C. Morel, G. Capodaglio, P. Cescon, G. Scarponi and C. Boutron, Environ. Sci. Technol., 2001, 35, 835 CrossRef CAS.
- C. Barbante, K. van de Velde, G. Cozzi, G. Capodaglio, P. Cescon, F. Planchon, S. Hong, C. Ferrari and C. Boutron, Environ. Sci. Technol., 2001, 35, 4026 CrossRef CAS.
- A. Matsumoto and T. K. Hinkley, Earth Planet. Sci. Lett., 2001, 186, 33 CrossRef CAS.
- J. R. McConnell, G. W. Lamorey and M. A. Hutterli, Geophys. Res. Lett., 2002, 29, 45-1 Search PubMed.
- J. R. McConnell, G. W. Lamorey, S. W. Lambert and K. C. Taylor, Environ. Sci. Technol., 2002, 36, 7 CrossRef CAS.
- C. Barbante, C. Boutron, A.-L. Moreau, C. Ferrari, K. van de Velde, G. Cozzi, C. Turetta and P. Cescon, J. Environ. Monit., 2002, 4, 960 RSC.
- C. Barbante, M. Swikowski, T. Döring, H. W. Gäggeler, U. Schotterer, L. Tobler, K. Van de Velde, C. Ferrari, G. Cozzi, A. Turetta, K. Rosman, M. Bolshov, G. Capodaglio, P. Cescon and C. Boutron, Environ. Sci. Technol., 2004, 38, 4085 CrossRef CAS.
- J. Zheng, C. Zdanowicz, D. Fisher, G. Hall and J. Vaive, J. Phys. IV, 2003, 107, 1405 Search PubMed.
- P. Gabrielli, F. A. M. Planchon, S. Hong, K. H. Lee, S. D. Hur, C. Barbante, C. P. Ferrari, J. R. Petit, V. Y. Lipenkov, P. Cescon and C. F. Boutron, Earth Planet. Sci. Lett., 2005, 234, 249 CrossRef CAS.
- K. H. Wedepohl, Geochim. Cosmochim. Acta, 1995, 59, 1217 CrossRef CAS.
- W. Shotyk, M. Krachler and B. Chen, in Anthropogenic Impacts on the Biogeochemistry and Cycling of Antimony in Metal Ions in Biological Systems, ed. A. Sigel, H. Sigel and R. K. O. Sigel, Taylor and Francis, Boca Raton, FL, vol. 44, pp.171–203 Search PubMed.
- US EPA, Water related fate of the 129 Priority Pollutants, Washington DC, 1979, Vol. 1 EP-440/4-79-029A.
- Council of European Union. Council Directive 98/83/EC of 3 November 1998, Quality of Water Intended for Human Consumption. Official Journal L 330, 05/12/1998, pp. 32–54.
- W. Shotyk, M. Krachler and B. Chen, Global Biogeochem. Cycles, 2004, 18 Search PubMed , GB1016:1.
- G. Weckwerth, Atmos. Environ., 2001, 35, 5525 CrossRef CAS.
- C. Dietl, W. Reifenhäuser and L. Peichl, Sci. Total Environ., 1997, 205, 235 CrossRef CAS.
- M. Krachler, M. Burow and H. Emons, J. Environ. Monit., 1999, 1, 477 RSC.
- J. O. Nriagu, Environment, 1990, 32, 8–28 Search PubMed.
- M. Krachler, J. Zheng, D. Fisher and W. Shotyk, Anal. Chem., 2004, 76, 5510 CrossRef CAS.
- M. Krachler, J. Zheng, D. Fischer and W. Shotyk, J. Anal. At. Spectrom., 2004, 19, 1017 RSC.
- M. Krachler, J. Zheng, D. Fischer and W. Shotyk, Anal. Chim. Acta, 2005, 530, 291 CrossRef CAS.
- http://thrust_2.tripod.com/glaciology/icedrill.html .
- J. Zheng, J. Vaive, M. Krachler, J. Lam, G. Lawson, G. Hall, C. Zdanowicz, D. Fisher and W. Shotyk, J. Environ. Monit., 2005 Search PubMed , submitted.
- C. Barbante, G. Cozzi, G. Capodaglio, K. van de Velde, C. Ferrari, C. Boutron and P. Cescon, J. Anal. At. Spectrom., 1999, 14, 1433 RSC.
- C. Barbante, T. Bellomi, G. Mezzadri, P. Cescon, G. Scarponi, C. Morel, S. Jay, K. Van de Velde, C. Ferrari and C. F. Boutron, J. Anal. At. Spectrom., 1997, 12, 925 RSC.
- J. M. Pacyna and E. G. Pacyna, Environ. Rev., 2001, 9, 269 CrossRef CAS.
- W. Shotyk, D. J. Weiss, J. D. Krames, R. Frei, A. K. Cheburbin, M. Gloor and S. Reese, Geochim. Cosmochim. Acta, 2001, 65, 2337 CrossRef CAS.
- K. A. Rahn, The Chemical Composition of the Atmospheric Aerosol, Technical report,University of Rhode Island, Kingston, RI, 1976 Search PubMed.
- W. Shotyk, M. Krachler, A. Martinez-Cortizas, A. K. Cheburkin and H. Emons, Earth Planet. Sci. Lett., 2002, 199, 21 CrossRef CAS.
- H. J. M. Bowen, Environmental Chemistry of the Elements, Academic Press, New York, 1979 Search PubMed.
- D. Stanners and P. Bourdeau, Europe’s Environment. The Dobris Assessment, European Environment Agency, Copenhagen, 1995 Search PubMed.
- R. Zevenhoven and P. Kilpinen, Control of Pollutants in Flue Gases and Fuel Gases, Helsinki University of Technology course ENE-47.153, ch. 8, ISBN 951-22-5527-8, available at http://users.tkk.fi/∼rzevenho/gasbook Search PubMed.
- N. Furuta, A. Iijima, A. Kambe, K. Sakai and K. Sato, J. Environ. Monit., 2005 10.1039/b513988k.
- W. T. Sturges, J. F. Hopper, L. A. Barrie and R. C. Schnell, Atmos. Environ., Part A, 1993, 27, 2865 CrossRef.
- W. T. Sturges and L. A. Barrie, Atmos. Environ., 1989, 23, 2513 CrossRef CAS.
- L. A. Barrie, Atmos. Environ., 1986, 20, 643 CrossRef CAS.
Footnote |
| † This work was presented at the First International Workshop on Antimony in the Environment, Heidelberg, Germany, 16th to 19th May 2005. |
| This journal is © The Royal Society of Chemistry 2005 |