Long-term relationships between SO2 and NOx emissions and SO42− and NO3− concentration in bulk deposition at the Hubbard Brook Experimental Forest, NH
Gene E.
Likens
*,
Donald C.
Buso
and
Thomas J.
Butler
Institute of Ecosystem Studies, P.O. Box AB, 65 Sharon Turnpike, Millbrook, New York, NY 12545, USA. E-mail: LikensG@ecostudies.org; Fax: +1-845-677-5976; Tel: +1-845-677-5343
First published on 8th August 2005
Abstract
A highly significant second-order polynomial relation between SO2 emissions and SO42− concentrations during 1970–2000 (r2 = 0.80, p = <0.001), and a linear relation between NOx and NO3− concentrations during 1991–2000 (r2 = 0.67, p = 0.004) in bulk precipitation were found for the Hubbard Brook Experimental Forest, NH based on emissions from a 24 h, back-trajectory determined source area. Earlier periods (1965–1980) for SO2 ∶ SO42− and longer periods (1965–2000) for NOx ∶ NO3− had poorer linear relations, r2 = 0.03, p = 0.51 and r2 = 0.22, p = 0.004, respectively. Methodology by the US Environmental Protection Agency for calculating emissions data during this period has changed significantly and frequently, making trend analysis difficult. Given the large potential for errors in estimating emissions and to a lesser extent, deposition, the robust relations between SO2 emissions and SO42− concentrations in bulk precipitation at the Hubbard Brook Experimental Forest show that careful, long-term measurements from a single monitoring site can provide sound and reasonable data on trends in air pollution.
1. Introduction
One of the most vexing questions in the US national debate about acid rain during the 1980’s was whether there was a clear relation between pollutant emissions (SO2 and NOx) and the resultant concentrations of SO42− and NO3− in precipitation downwind of the emission sources. This question had clear and important policy and ecological implications, but neither long-term nor experimental data were available at that time to provide a clear answer.Long-term data on precipitation chemistry from 1965 to the present from the Hubbard Brook Experimental Forest (HBEF) in the White Mountains of New Hampshire are now available and can be used to address this problem. Our attempts to correlate anthropogenic emissions of SO2 and NOx from source areas impacting the HBEF, with long-term measurements of SO42− and NO3− in bulk precipitation produced some surprising results. We present and discuss these results here.
2. Site
The HBEF is located in central New Hampshire (43°56′ N, 71°45′ W) USA. Climate is humid, continental with short, cool summers and long, cold winters. Since 1963–64, annual precipitation has averaged about 142 cm, with 25–33% falling as snow (Federer et al.;1 A. Bailey et al.2). Soil, vegetation, topographic, geologic and other ecologic characteristics are described in Likens and Bormann.33. Procedures
Volume-weighted, annual (calendar-year) concentrations of SO42−and NO3− are used here to correlate with annual emissions of SO2 and NOx from a prescribed emissions source area for the HBEF. Samples of bulk deposition have been collected and analyzed for chemical content on a weekly basis at the HBEF since 1963–64 with a continuously open plastic funnel and reservoir system for rain and a plastic garbage can/bucket system for snow. A detailed description and analysis of this methodology is given in Likens and Bormann3 and Buso et al.4 The annual error in measuring precipitation volume is ∼5% and for chemical analyses is ∼5% (Buso et al.;4 S. Bailey et al.5).Annual SO2 and NOx emission values were obtained from the EPA Emissions Factor and Inventory Group (EPA6–8). Emissions data for individual states were converted from short tons of SO2 and NOx year−1 into metric tons year−1. In addition, we obtained Canadian SO2 and NOx emissions at the provincial level, as well as emission densities within provinces from Environment Canada.9,10
To establish the emission source regions for the HBEF we used the NOAA Air Resources Laboratory, Hysplit-4 model (Draxler and Hess11) to define air-mass, back trajectories starting at 500 m above ground level (agl) and also at 1000 m agl. Trajectories at 500 m agl are within the boundary layer and at 1000 m agl should be near the top of the boundary layer. The lower 500 m, air-mass, back trajectories at times show a more easterly component, representing surface air drawn into an approaching frontal system. The back trajectories were calculated for two years (1995 and 1996) on days when at least 2.5 mm of precipitation occurred (n = 167). Using cluster analysis (Stunder12) we obtained representative mean back trajectories for both years combined. Previous work (Butler et al.13), which compared up to four individual years of back trajectories, has shown that source regions do not change significantly from year to year in the eastern USA.
Using cluster analysis and summary plots of all the wet-day, back trajectories for 1995 and 1996, we established three, different-sized source areas for the HBEF based on 12 h, 24 h and 36 h back-trajectory periods (Fig. 1). In previous work (Likens et al.;14,15 Butler et al.13), 15 h back trajectories starting at 2000 m agl were used to estimate the appropriate source regions that could impact the HBEF area. The patterns of emissions for both the 15 h (start height = 2000 m agl) and 24 h (start heights = 500 m and 1000 m agl) were very similar (Fig. 2). The correlation coefficient (r) values for relating the current emissions estimates to previous estimates were all >0.99 (p = <0.0001).
 | ||
| Fig. 1 Emission source areas for the HBEF based on 12 h (lightest areas adjacent to HBEF), 24 h (lightest areas plus darkest areas) and 36 h (lightest, darkest and mid-grey areas) air-mass back trajectories. | ||
![Emission data for SO2
(A) and NOx
(B) for the Hubbard Brook Experimental Forest from 1940 to 2000 based on different criteria used by the Environmental Protection Agency (EPA)
[see text]: –○– , most recent data (received from the EPA Emission Factor and Inventory Group on 3 December 2004) for the HBEF, 15 h back-trajectory, source area; –●– , previous EPA data for the HBEF, 15 h back-trajectory, source area, SO2 data published in Likens et al.;15
–▲– , updated EPA data for the HBEF, 24 h back-trajectory, source area, NOx data used in Butler et al.16 The 15 h source area has a back trajectory start height of 2000 m agl. The 24 h source area has back trajectory start heights of 500 m and 1000 m agl. The sharp decline in SO2 from 1994 to 1995 is the result of the initial implementation of the 1990 Clean Air Act Amendments. Canadian emissions are not included in this data set in order to focus on EPA emissions data. The r values for relating the most recent emissions estimates to previous estimates are all >0.99 (p
= <0.0001).](/image/article/2005/EM/b506370a/b506370a-f2.gif) | ||
| Fig. 2 Emission data for SO2 (A) and NOx (B) for the Hubbard Brook Experimental Forest from 1940 to 2000 based on different criteria used by the Environmental Protection Agency (EPA) [see text]: –○– , most recent data (received from the EPA Emission Factor and Inventory Group on 3 December 2004) for the HBEF, 15 h back-trajectory, source area; –●– , previous EPA data for the HBEF, 15 h back-trajectory, source area, SO2 data published in Likens et al.;15 –▲– , updated EPA data for the HBEF, 24 h back-trajectory, source area, NOx data used in Butler et al.16 The 15 h source area has a back trajectory start height of 2000 m agl. The 24 h source area has back trajectory start heights of 500 m and 1000 m agl. The sharp decline in SO2 from 1994 to 1995 is the result of the initial implementation of the 1990 Clean Air Act Amendments. Canadian emissions are not included in this data set in order to focus on EPA emissions data. The r values for relating the most recent emissions estimates to previous estimates are all >0.99 (p = <0.0001). | ||
Canadian emissions from the provinces of Ontario and Quebec were also included in the source regions. The record we obtained for Canadian emissions only extended back to 1980. However, we estimated Canadian emissions for the years previous to 1980, by taking the mean % of total emissions that were from Canadian sources for the period 1980 to 1984 and applying this percentage of emissions to the previous years. For 1980–1984, Canadian emissions from Ontario and Quebec accounted for 25% of the total SO2 emissions and 13% of the NOx emissions for the 24 h source regions pertinent to the HBEF. In the latter part of the record (1996 to 2000), Canadian SO2 and NOx emissions represented 14% and 13% of the total emissions, respectively. Thus, Canadian SO2 emissions showed a decline in relative importance, while Canadian NOx emissions maintained the same relative importance during the latter part of the record for emissions. More discussion regarding establishment of source regions can be found in Butler et al.16
In our attempt to obtain and use Environmental Protection Agency (EPA) data on emissions of SO2 and NOx, we discovered some problems with these long-term data. The quality of the emissions data and how they were estimated have been variable during the period of this record. The 1963 to 2000 emissions data were based on two records of emissions: the SO2 and NOx emissions from the “archive years”, 1900 to 1984, and the “current years”, 1985 to 2000 (EPA17). The emissions data for the “archive years” were based on a top-down approach, where national information and emission factors were used to create estimates of national emissions that were disaggregated to the state level. The methodology during the last several years was revised periodically by the EPA for the “current years” (1985 to 2000), during which an opposite, bottom-up, approach was applied. That is, emissions were derived at the county level to incorporate more state or local agency emissions data (EPA17).
Both the NOx and SO2 record of emissions for the “archive” and “current” years overlapped for the period 1985 to 1994. Data during these overlapping years were essentially the same until 1990; thereafter, the “current” emissions for both NOx and SO2 were higher than the data based on “archive” emissions (Fig. 1). A spike in emissions from 1989 to 1990, occurred for most states. This change happened at the same time that there was a change in the methodology (EPA18) and probably is, at least in part, an artifact of the methodology change. Despite this potential artifact, we used the record for “current” years of state level data as it was presented by the EPA for the years 1985 to 2000 (EPA6–8). It should be noted that more recent estimates of emissions can change, depending on which EPA trend report is used. For example, data from the EPA National Air Quality and Emissions Trends Report Special Studies Edition,19 differ when compared with national EPA emissions data released in 2005 (EPA20). Annual differences in national SO2 and NOx emissions were 2 to 11% and 0 to 11%, respectively, for the period 1990–2000. The different SO2 emission data showed very similar trends (r = 0.99, p = <0.001), however, the trends for NOx emissions were in the opposite direction (from 1990 to 1997) and the annual NOx emissions from the two sources were poorly correlated (r = −0.20, p = 0.55) and statistically not significant.
There is a further complication regarding estimates of NOx emissions. A recent assessment of EPA emission estimates (NARSTO21) calls into question the accuracy of US highway-vehicle NOx estimates. For the 24 h HBEF source area, vehicle emissions represented 52% of the total, and highway vehicle NOx emissions represented 35% of the total NOx emissions during this period. In the NARSTO assessment of emissions, results of a top-down analysis (as compared to the EPA’s bottom-up approach) showed an increasing trend in highway vehicle emissions of 1.9% per year for the period 1990 to 2000, whereas national EPA estimates of highway vehicle emissions showed a declining trend of 1.1% per year for the same period. The uncertainty associated with highway vehicle estimates may also apply to off-highway vehicles, which clearly points to the need for the EPA to develop more accurate NOx emissions data.
5. Results
Clearly, the different methodology used by the EPA in calculating emissions of SO2 and NOx has produced some different annual values in the latter part of the record; i.e. since 1940 (Fig. 2). We also have compared different-sized source areas for emissions (Fig. 3), since airshed boundaries cannot be defined easily. As might be expected, greater amounts of emissions are produced from larger source areas. Annual emissions are about 4× and 3× greater from 36 h back trajectories than from 12 h back trajectories for SO2 and NOx, respectively. However, the trends in emissions are quite similar (Fig. 3). For SO2 emissions, the r values were 0.96, (p = <0.0001) and >0.99 (p = <0.0001) when comparing the 24 h source region to the 12 and 36 h source regions, respectively. For NOx emissions, the r values were 0.97 (p = <0.0001) and 0.98 (p = <0.0001) for the same comparisons. | ||
| Fig. 3 Emission data for SO2 and NOx for the Hubbard Brook Experimental Forest from 1985 to 2000 based on updated EPA plus Canadian data for 12 h (–○–), 24 h (–●–) and 36 h (–▲–) back-trajectory, source areas. For SO2, linear regression trends were all significantly negative (p < 0.001): 12 h, r2 = 0.88; 24 h, r2 = 0.84; 36 h, r2 = 0.81. | ||
The long-term relation between SO2 emissions from the 24 h source area and the SO42− concentration in bulk precipitation for the HBEF is shown in Fig. 4A. Using the long-term data for the HBEF, the curvilinear relation between annual SO2 emissions and SO42− concentration has become quite strong (r2 = 0.80, p = <0.001; Fig. 4A), which is remarkable given all the inherent variability in meteorology, methodology, etc. that is embedded in this relationship. During 1965–1969, the slope was negative (−6.3X + 84.1; r2 = 0.86, p = 0.023). A weaker relation existed between NOx and NO3− concentrations for the period 1965 to 2000 for the Hubbard Brook Experimental Forest (r2 = 0.22, p = 0.004). However, for the period 1991 to 2000, when greater changes in NOx emissions occurred, the relation became more significant (r2 = 0.69, p = 0.003) (Fig. 4B) for the data that included a “correction” for highway vehicle emissions. If no correction was applied, the r2 was 0.67 (p = 0.004).
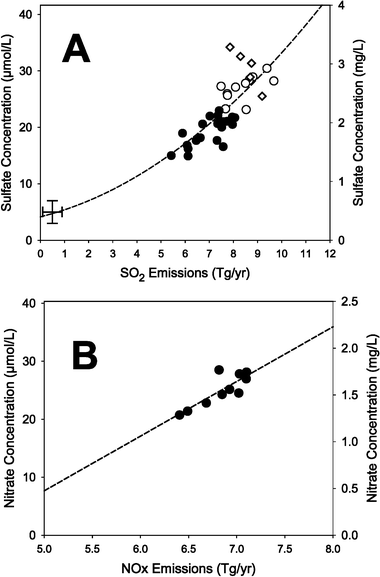 | ||
Fig. 4 Relationships between (A) updated SO2 emissions (24 h source area) and SO42− concentrations in bulk precipitation during 1965–2000, and (B) NOx emissions (24 h source area) and NO3− concentrations in bulk precipitation for the Hubbard Brook Experimental Forest during 1991–2000. These analyses include Canadian emissions from Ontario and Quebec. Panel A: ◇
= 1965–1969; ○
= 1970–1980; ●
= 1981–2000. The second-order polynomial regression including the PIR point for the SO2
∶ SO42− relation during 1970–2000 has an r2
= 0.80 (p
= <0.0001).  is an estimate of values for the Pre-Industrial Revolution period. Panel B: The NOx emissions include a 1.9% per year increase for NOx from vehicle emissions derived from NARSTO.21 The linear regression for NOx
∶ NO3− during 1991–2000 has an r2
= 0.69 (p
= 0.003). If the 1.9% per year vehicle emissions correction is not used, the r2
= 0.67 (p
= 0.004). is an estimate of values for the Pre-Industrial Revolution period. Panel B: The NOx emissions include a 1.9% per year increase for NOx from vehicle emissions derived from NARSTO.21 The linear regression for NOx
∶ NO3− during 1991–2000 has an r2
= 0.69 (p
= 0.003). If the 1.9% per year vehicle emissions correction is not used, the r2
= 0.67 (p
= 0.004). | ||
6. Discussion
We are interested in the results of trend analyses for the ever-lengthening record of precipitation chemistry at the HBEF (now more than four decades). Relating these data to emissions is difficult, however, because the EPA has regularly changed its methodology, etc. for deriving emission data for SO2 and NOx. For example, the EPA has stated: “In the past, the emission estimates presented in the ‘Trends’ reports would change from one year to the next based on the development of new information, data, or methodologies used to estimate the emissions. These changes were applied not only to the most recent year, but to all or some of the preceding years.”18 Thus, our previously published efforts to show these relations14,15 are now out-of-date or in question as the data on emissions have been changed retrospectively, creating a moving target. Here, we have attempted to provide updated information about these relations, especially since the EPA has stated, “As of 1997, no such changes are planned to be made to the emissions for the years prior to 1985.”18There is an important need to answer the politically and ecologically important questions relative to federal and state-mandated regulations to reduce emissions of SO2 and NOx: “Do the reduced emissions make any difference, and are we getting our money’s worth from expensive legislation?” Clearly, the long-term data from HBEF show that reducing emissions of SO2 decreases the concentration of SO42− in bulk precipitation. As a result, the answer to both questions is, “yes” for sulfur. The answers regarding changes in NOx emissions from 1991 to 2000 also appears to be “yes”, with the caveat that higher quality NOx emissions data, especially vehicle emissions, are necessary to quantify more reliably the relation between changing NOx emissions and changing NO3− concentrations in precipitation. Also, reductions in total emissions of NOx have been relatively small until recently (Butler et al.16), and thus, relations between NOx emissions and NO3− in precipitation are more difficult to discern.
Such results have clear policy and management implications—what goes up into the atmosphere as emissions, directly influences what comes down in precipitation. In Fig. 4A, we show this relationship for concentrations of SO42− in bulk precipitation at HBEF. Obviously, inputs to ecosystems are based on concentration × amount of precipitation, and amount of precipitation can be highly variable throughout landscapes and regions. Nevertheless, predicting deposition for important nutrients and/or pollutants is critical for understanding ecological impact and for the conservation of ecosystems.
The strong relation between SO2 emissions and SO42− concentrations in bulk precipitation for the HBEF shown in Fig. 4A does not include estimates for dry components (SO2 and particulate sulfate) in deposition. Assuming that dry deposition is 21% of bulk deposition for the HBEF (Likens et al.15), we can estimate roughly the proportion of total sulfur emissions from the 24 h source area that has been deposited on the entire HBEF during 1965–2000. It is <0.001%! That a strong relationship exists between changing SO2 emissions from the source area and SO42− concentration in bulk deposition despite the very small proportion of emissions that are actually deposited on the HBEF is indicative of the remarkable homogeneity of mixing during long-range transport in the atmosphere.
Long-term data to examine the relation between emissions and deposition are rare, but more recent, shorter-term records from other sites in New York and New England show relationships for SO2 ∶ SO42− similar to what we have reported here (Butler et al.;13 Driscoll et al.23).
The value of the long-term data became very clear in this analysis. Based on calendar-year data, no significant relationship was apparent between SO2 emissions and SO42− concentrations in bulk precipitation at the HBEF (24 h source area) during 1965–1980 (r2 = 0.03, p = 0.51), so it is not surprising that the debate over links between sulfur emissions and deposition occurred during that period. With a longer record (1970–2000), however, the curvilinear relationship became robust (Fig. 4A; r2 = 0.80, p = <0.001). A significant, but not strong relation was apparent between NOx emissions and NO3− concentration in precipitation at the HBEF for the period 1965 to 2000 (r2 = 0.22, p = 0.004). One reason for this poorer correlation is the high degree of year-to-year variability in the concentration of NO3− in precipitation. The mean between-year % difference is 14% (σ = ±12%), but the % difference in some years is as high as 50%. However, despite these variations, with improved emission estimates, and a greater change in NOx emissions per year a significant relation exists between NOx emissions and NO3− concentration (r2 = 0.69, p = 0.003) for the period 1991 to 2000 (Fig. 4B). The period after 1990 also had the most consistency in how NOx emissions were calculated (EPA18).
Although it may not be determined why the relation between SO2 and SO42− was so variable from 1964 to 1980 at the HBEF, we can speculate on some possible explanations. The period 1964–66 was a time of severe drought in the northeastern US (Likens and Bormann3) affecting meteorologic patterns of air mass trajectories and atmospheric deposition. In addition, during 1964–70 there was an increase in emissions from local and distant sources (e.g., the copper-nickel smelter in Sudbury, Ontario), and increases in stack heights (Likens22). There were greater uncertainties in the measurements of bulk precipitation (Buso et al.4) and particularly emissions (see above).
7. Conclusions
A clear and strong relation between SO2 emissions from the 24 h source area and SO42− concentrations in bulk precipitation for the HBEF was shown in long-term data (1965–2000). Sulfate concentration data from 1965–1980, however, showed an insignificant relation (r2 = 0.03, p = 0.51) with SO2 emissions from the 24 h, back-trajectory source area.A weak statistical relation (r2 = 0.22, p = 0.004) was apparent for EPA emissions of NOx and concentrations of NO3− at HBEF for the long-term record (1965–2000), however, no statistical relation was evident with data from 1985–2000 (r2 = 0.17, p = 0.11). A good relation between NOx emissions, including Canadian emissions and a correction for vehicular emission and concentrations of NO3−, was shown for the period 1991–2000 (r2 = 0.69, p = 0.003).
Changes in methodology by the EPA in calculating emission data for SO2 and NOx have made trend analyses difficult and a moving target, and a recent assessment indicates that the highway vehicle NOx emission estimates by the EPA are not correct.
On average, only <0.001% of total S emissions from the 24 h, back-trajectory source area were estimated to be deposited on the entire HBEF annually during 1965–2000. Nevertheless, the relationship between emissions and precipitation concentration was robust, demonstrating that careful, long-term measurements from a single, deposition collection site provide a sound and reasonable approach for monitoring trends in air pollution.
Acknowledgements
Financial support was provided by The Andrew W. Mellon Foundation and the National Science Foundation. State level data on SO2 and NOx emissions were provided by Thomas McMullen of the EPA Emissions Factor and Inventory Group. Barbara Stunder provided assistance with back trajectory analysis used in estimating emission source areas. We thank Carolyn Klocker for help in preparing the source area map and P. C. Likens for manuscript preparation. The Hubbard Brook Experimental Forest is operated and maintained by the USDA Forest Service, Newtown Square, PA. This is a contribution to the Hubbard Brook Ecosystem Study and to the program of the Institute of Ecosystem Studies. This publication does not reflect the view of any sponsoring agency.References
- C. A. Federer, L. D. Flynn, C. W. Martin, J. W. Hornbeck and R. S. Pierce, Thirty years of hydrometeorologic data at the Hubbard Brook Experimental Forest, New Hampshire, General Technical Report NE-141, USDA Forest Service, 1990, p. 44 Search PubMed.
- A. S. Bailey, J. W. Hornbeck, J. L. Campbell and C. Eagar, Hydrometeorological database for Hubbard Brook Experimental Forest: 1955–2000, Gen. Tech. Report NE-305, USDA Forest Service, Northeastern Research Station, 2003, p. 36 Search PubMed.
- G. E. Likens and F. H. Bormann, Biogeochemistry of a Forested Ecosystem, Springer-Verlag, New York Inc., 2nd edn., 1995, p. 159 Search PubMed.
- D. C. Buso, G. E. Likens and J. S. Eaton, Chemistry of precipitation streamwater and lakewater from the Hubbard Brook Ecosystem Study: A record of sampling protocols and analytical procedures, General Tech. Report NE-275, USDA Forest Service, Northeastern Research Station, Newtown Square, PA, 2000, p. 52 Search PubMed.
- S. W. Bailey, D. C. Buso and G. E. Likens, Ecology, 2003, 84, 471 Search PubMed.
- EPA, National Emissions Inventory (NEI), Emissions Factor Inventory Group, Office of Air Quality Planning and Standards, US EPA, Research Triangle Park, NC 27711, USA, 2001a Search PubMed.
- EPA, National Emissions Inventory (NEI), Emissions Factor Inventory Group, Office of Air Quality Planning and Standards, US EPA, Research Triangle Park, NC 27711, USA, 2002 Search PubMed.
- EPA, State level emissions data obtained from Emission Factor and Inventory Group, US EPA, Research Triangle Park, NC 27711, USA, December 2004 Search PubMed.
- Environment Canada, Data from 1990–1994, and 1996–1998, in Annual Reports on the Federal/Provincial Agreements for the Eastern Canada Acid Rain Program, 1990 to 1998, Pollution Data Branch, Environmental Protection Service, Environment Canada, Ottawa, Ontario Search PubMed.
- (a) 2000 Annual Progress Report on the Canada-Wide Acid Rain Strategy for Post 2000 (data for 1995, 1999 and 2000), Environment Canada, 2001a Search PubMed; (b) Environment Canada CAC Emission Summaries at http://www.ec.gc.ca/pdb/ape/cape_tables/nox95_e.cfm, Environment Canada, 2001b; (c) Environment Canada Air Pollutant Emissions at http://www.ec.gc.ca/pdb/ape/cape_home_e.cfm and provincial emission density maps at http://www.ec.gc.ca/pdb/ape/cape/maps/cenox_e.cfm, Environment Canada, 2001c.
- R. R. Draxler and G. D. Hess, Aust. Meteorol. Mag., 1998, 47, 295 Search PubMed.
- B. J. B. Stunder, J. Appl. Meteorol., 1996, 35, 1319 Search PubMed.
- T. J. Butler, G. E. Likens and B. J. B. Stunder, Atmos. Environ., 2001, 35, 1015 CrossRef CAS.
- G. E. Likens, T. J. Butler and D. C. Buso, Biogeochemistry, 2001, 52, 1 CrossRef CAS.
- G. E. Likens, C. T. Driscoll, D. C. Buso, M. J. Mitchell, G. M. Lovett, S. W. Bailey, T. Siccama, W. A. Reiners and C. Alewell, Biogeochemistry, 2002, 60, 235 CrossRef CAS.
- T. J. Butler, G. E. Likens, F. M. Vermeylen and B. J. B. Stunder, Atmos. Environ., 2003, 37, 2093 CrossRef CAS.
- EPA, Procedures Document for National Emission Inventory, Criteria Air Pollutants 1985–1999, EPA-454/R-01-006, 2001b, p. 409 Search PubMed.
- EPA, National Air Pollutant Emission Trends, Procedures Document, 1900–1996, EPA-454/R-98-008, US EPA, Research Triangle Park, NC 27711, USA, 1998, p. 291 Search PubMed.
- EPA, National Air Quality and Emissions Trends Report, Special Studies Edition, EPA 454/R-03-005 (http://www.epa.gov/air/airtrends/aqtrnd03), US EPA, Research Triangle Park, NC 27711, USA, 2003 Search PubMed.
- EPA, 1970–2002 Average annual emissions, all criteria pollutants Jan. 2005 (http://www.epa.gov/tnn/chief/trends/index.html), US EPA, Research Triangle Park, NC 27711, USA, 2005 Search PubMed.
- NARSTO (North American Research Strategy for Tropospheric Ozone), Improving Emission Inventories for Effective Air Quality Management across North America—A NARSTO Assessment, http://www.cgenv.com/narsto/ Search PubMed.
- G. E. Likens, Ind. Crisis Q., 1987, 1, 13 Search PubMed.
- C. T. Driscoll, K. M. Driscoll, M. J. Mitchell and D. J. Raynal, Environ. Pollut., 2003, 123, 327 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2005 |