Thio arsenosugars in freshwater mussels from the Danube in Hungary
Csilla
Soeroes
*a,
Walter
Goessler
b,
Kevin A.
Francesconi
b,
Ernst
Schmeisser
b,
Reingard
Raml
b,
Norbert
Kienzl
b,
Markus
Kahn
b,
Peter
Fodor
a and
Doris
Kuehnelt
b
aDepartment of Applied Chemistry, Faculty of Food Science, Corvinus University, Villányi út 29-33, 1118 Budapest, Hungary. E-mail: csilla.soros@uni-corvinus.hu; Fax: +36 1 466 4272; Tel: +36 1 482 6166
bInstitute of Chemistry—Analytical Chemistry, Karl-Franzens University Graz, Universitaetsplatz 1, 8010 Graz, Austria
First published on 10th June 2005
Abstract
In contrast to the large body of data on naturally-occurring arsenic compounds in marine organisms, relatively little is known about arsenic speciation in freshwater biota. We report an investigation using HPLC-ICPMS into the arsenic compounds in five species of freshwater mussels collected from five sites from the Danube in Hungary. Total arsenic concentrations in the mussels ranged from 3.8–12.8 mg As kg−1. The arsenic speciation patterns were broadly similar for mussels representing each of the five species and five sites, but quite different from those reported for marine mussels. The major extractable arsenicals were two oxo arsenosugars (glycerol sugar and phosphate sugar), and their thio analogues (thio glycerol sugar and thio phosphate sugar). Arsenobetaine, usually the major arsenical in marine organisms, was not a significant compound in the freshwater mussels and was detected in only three of the 11 samples. This is the first report of thio arsenosugars in freshwater biota and suggests that these compounds may be common and widespread naturally-occurring arsenicals.
Introduction
Mussels are very important inhabitants of the freshwater ecosystem. As primary consumers, they occupy an intermediate position in the food web, shunting energy from one trophic level to another. Their food is obtained through filter feeding; phytoplankton, small zooplankton, bacteria and detritus make up the bulk of their feed. These “living filters” are known to be able to accumulate metals and metalloids in their soft body parts, making them suitable for biomonitoring purposes.1,2 As mussels are prey for many types of fish and small mammals, the accumulated metals and metalloids can be transferred through the food web, and ultimately reach humans.Marine mussels naturally contain arsenic at the mg kg−1 level and the major arsenic form is arsenobetaine (AB, Fig. 1), a compound which is interpreted to be of low toxicity and seems to be the end-point of the biotransformation of arsenic in marine ecosystems.3,4
 | ||
| Fig. 1 Structures of arsenic compounds relevant to this work (compounds are drawn in their most deprotonated form). | ||
Total arsenic concentrations in freshwater mussels are comparable with those in marine mussels, but they are considerably higher than the concentrations reported for other freshwater biota from non-polluted sites.5 Because mussels incorporate higher concentrations of metalloids than other freshwater organisms, they are very suitable organisms for an arsenic speciation study in a freshwater system.
Little is known about arsenic speciation in freshwater food-chains, especially in mussels.6 Most of the reported work has been based on samples from arsenic-rich waters or on artificially exposed biota. However, these ‘forced’ circumstances may never completely and accurately reflect the real natural biochemical processes. There have been just two previous studies of arsenic species in freshwater mussels. Slejkovec et al.7 found that 51% of the extractable arsenic in Dreissena polymorpha was present as AB and/or TMAO (the method applied was not able to distinguish between these two arsenic compounds). On the other hand, Koch et al.5 found that an extract of Margaritifera sp. contained mainly DMA and arsenosugars (Fig. 1), and that AB did not occur. Those researchers investigated Anadonta sp. as well, which also contained arsenosugars, but no AB, in addition to some As(V) and an unknown arsenic compound.
The present study aims to expand our knowledge of arsenic in freshwater ecosystems by examining total arsenic concentrations and arsenic compounds in the soft-body of specimens representing five mussel species collected from the Danube in Hungary, a natural river system. Mussels from the Unionidae family were used for this purpose because they are abundant in the river Danube and are ecologically important inhabitants of shallow water areas.
Materials and methods
Sample collection and sample preparation
Five mussel species (Unio tumidus, Unio pictorum, Anadonta anatina, Sinanadonta woodiana and Dreissena polymorpha) were collected from five sampling points on the river Danube in April 1999 (Table 1). The five sampling locations were selected to cover the whole Danube region in Hungary. The molluscs were collected by hand in depths of 0.2–2 m by snorkelling and scuba diving. They were then transported to the laboratory in tanks filled with ambient water, where they were kept for a maximum of 24 h. The animals were then stored whole in a freezer (−20 °C) until their analysis in 2004. After thawing the samples, the lengths of the individuals were measured and the whole soft-body tissue was rinsed with deionized water. Samples from the same sampling site and same biological species were pooled resulting in 11 groups of samples (Table 1); they were then deep-frozen, and freeze-dried in a Christ Alpha 1–4 freeze-drying system (Christ, Osterode am Harz, Germany). The dry samples were pulverized and homogenized in a laboratory porcelain mortar by hand.| Mussel species | Sampling place | Average size of the mussels/cm (RSD%) n = number of individuals | Total As concentration/μg As kg−1 dry masse | ABa | DMAb | As(V)c | Glycerol sugara | Phosphate sugarb | Thio glycerol sugarcd | Thio phosphate sugarcd | Sum of species |
|---|---|---|---|---|---|---|---|---|---|---|---|
| /μg As kg−1 dry mass (% of the summed species) | |||||||||||
| a Determined with the ZORBAX 300-SCX column at pH 2.6; d.l.: detection limit (10 μg As kg−1). b Determined with the HAMILTON PRP-X100 column at pH 5.6. c Determined with the HAMILTON PRP-X100 column at pH 10.3. d Quantified with the calibration curve for As(V). e Pooled sample analysed in triplicate (RSDs ≤ 8.5%, mean RSD 3.8%). | |||||||||||
| Anadonta anatina | Dunafajsz | 8.3 (26) | 5180 | 16.3 | 41.4 | 28.1 | 248 | 216 | 99.4 | 142 | 791 |
| n = 3 | (2.1) | (5.2) | (3.6) | (31.3) | (27.3) | (12.6) | (17.9) | ||||
| Anadonta anatina | Dunamedve | 7.9 (6) | 4150 | <d.l. | 39.0 | 25.9 | 145 | 210 | 107 | 147 | 674 |
| n = 4 | (5.8) | (3.8) | (21.5) | (31.2) | (15.9) | (21.8) | |||||
| Anadonta anatine | Vág | 7.2 (20) | 4680 | 46.1 | 35.4 | 44.3 | 353 | 273 | 142 | 158 | 1052 |
| n = 4 | (4.4) | (3.4) | (4.2) | (33.6) | (26.0) | (13.5) | (15.0) | ||||
| Dreissena polymorpha | Dunafajsz | <0.8 | 4500 | <d.l. | 39.6 | 69.3 | 194 | 182 | 79.9 | 89.8 | 655 |
| n = 25 | (6.0) | (10.6) | (29.6) | (27.8) | (12.2) | (13.7) | |||||
| Sinanadonta woodiana | Dunaföldvár | 9.9 (19) | 5140 | <d.l. | 96.6 | 11.3 | 192 | 404 | 58.1 | 133 | 895 |
| n = 4 | (10.8) | (1.3) | (21.5) | (45.1) | (6.5) | (14.9) | |||||
| Sinanadonta woodiana | Dunafajsz | 14.0 | 3800 | <d.l. | 41.1 | Trace | 72.5 | 180 | 25.0 | 86.9 | 406 |
| n = 1 | (10.1) | (17.9) | (44.4) | (6.2) | (21.4) | ||||||
| Unio pictorum | Dunafajsz | 9.0 (20) | 6850 | Trace | 52.7 | 18.5 | 356 | 397 | 100 | 176 | 1100 |
| n = 3 | (4.8) | (1.7) | (32.4) | (36.1) | (9.1) | (16.0) | |||||
| Unio pictorum | Dunaföldvár | 10.4 (10) | 12770 | <d.l. | 93.2 | Trace | 678 | 503 | 195 | 235 | 1704 |
| n = 3 | (5.5) | (39.8) | (29.5) | (11.4) | (13.8) | ||||||
| Unio pictorum | Vác | 10.3 (12) | 8860 | <d.l. | 92.1 | 15.6 | 701 | 614 | 179 | 171 | 1773 |
| n = 3 | (5.2) | (0.9) | (39.5) | (34.6) | (10.1) | (9.6) | |||||
| Unio tumidus | Vác | 9.2 (12) | 6530 | <d.l. | 52.5 | Trace | 329 | 284 | 91.7 | 85.5 | 843 |
| n = 3 | (6.2) | (39.0) | (33.7) | (10.9) | (10.1) | ||||||
| Unio tumidus | Vág | 8.6 (14) | 5960 | <d.l. | 56.1 | 15.9 | 323 | 247 | 153 | 196 | 991 |
| n = 3 | (5.7) | (1.6) | (32.6) | (24.9) | (15.4) | (19.8) | |||||
Instrumentation
Digestions for total arsenic determinations were performed with a Milestone ultraCLAVE II microwave digestion system (EMLS, Leutkirch, Germany). Total arsenic determinations and arsenic speciation analyses were carried out with an Agilent 7500c inductively coupled plasma mass spectrometer (ICPMS) (Agilent, Waldbronn, Germany) as detection system. An Agilent 1100 Series HPLC system (Agilent, Waldbronn, Germany) consisting of a solvent degassing unit, a binary pump, an autosampler, and a thermostatted column compartment was used as the chromatographic system. The chromatographic conditions applied are summarized in Table 2. The analytical columns were protected by guard columns filled with the same stationary phases. The outlet of the HPLC column was connected via PEEK capillary tubing (0.125 mm i.d.) to the nebulizer of the ICPMS system, which served as the arsenic selective detector. The ion intensity at m/z 75 (75As) was monitored using the ‘time-resolved’ analysis software. Additionally, the ion intensities at m/z 77 (40Ar37Cl and 77Se) and 82 (82Se) were monitored to detect possible argon chloride (40Ar35Cl) interferences on m/z 75. Instrumental settings used throughout this work were described in detail elsewhere.8 Peak areas were determined using the ICPMS chromatographic software version C.01.00 (Agilent, Waldbronn, Germany).| Chromatographic condition I cation-exchange | Chromatographic condition II anion-exchange12 | Chromatographic condition III anion-exchange13 | |
|---|---|---|---|
| Column | ZORBAX 300-SCX 15 cm × 4.6 mm, 5 μm (Agilent, Waldbronn Germany) | PRP-X100 25 cm × 4.1 mm, 10 μm (Hamilton, Reno, USA) | PRP-X100 10 cm × 4.1 mm, 5 μm (Hamilton, Reno, USA) |
| Mobile phase | 10 mM pyridine | 20 mM NH4H2PO4 | 20 mM NH4HCO3 |
| pH | 2.6 (adjusted with formic acid) | 5.6 (adjusted with 25% aqueous NH3) | 10.3 (adjusted with 25% aqueous NH3) |
| Injection volume/mm3 | 20 | 20 | 20 |
| Column temperature/°C | 30 | 40 | 40 |
| Flow rate/cm3 min−1 | 1.5 | 1.5 | 1.5 |
| Quantified arsenic species | AB, glycerol sugar, TMAO, AC, TETRA | DMA, MA, phosphate sugar, As(V), sulfonate sugar, sulfate sugar | As(V), thio glycerol sugar, thio phosphate sugar |
Reagents, standards and reference materials
All solutions were prepared with Milli-Q (18.2 MΩ cm) water. Concentrated nitric acid (Merck, puriss analytical—p.a.) was further purified in a quartz sub-boiling distillation unit. Methanol (puriss p.a.), formic acid (puriss p.a.), ammonium hydrogen carbonate and ammonium dihydrogen phosphate (p.a.) were purchased from Fluka (Buchs, Switzerland), and pyridine (p.a.) and Na2HAsO4·7H2O from Merck (Darmstadt, Germany).Standard solutions (1000 μg As cm−3) for the identification and quantification of arsenic compounds were prepared as described elsewhere.8 Arsenosugars were isolated from natural sources and purified as described elsewhere.9 The synthesis of the thio glycerol and thio phosphate sugars was carried out by bubbling H2S through aqueous solutions of glycerol sugar (3 μg As cm−3) or phosphate sugar (1 μg As cm−3). The stock solutions were diluted with water to the desired concentrations just before use.
For total arsenic determinations, 72Ge (50 ng cm−3) was used as internal standard. Quality control of the total arsenic determinations in the mussel samples was performed by the analysis of the certified reference material DOLT-2 (dogfish liver, National Research Council Canada, Ottawa, Canada).
Determination of total arsenic
Total arsenic was determined by ICPMS after decomposition of the mussel samples with HNO3 in the microwave digestion device. Portions of the samples (0.3 g) or DOLT-2 (0.2 g) were weighed into 12 cm3 quartz vials and concentrated nitric acid (5 cm3) was added. The quartz vials were then closed with Teflon® caps and placed into the sample rack. After loading the autoclave with argon to a pressure of 40 bar, the samples were heated to 250 °C and held at this temperature for 30 min. The digests were filled to 50 cm3 with Milli-Q water. An external calibration curve established with As(V) solutions was used for quantification. Each of the eleven pooled samples was analysed in triplicate; RSDs were ≤8.5% (mean = 3.8%). Analysis of the certified reference material DOLT-2 (16.6 ± 1.1 mg kg−1) gave a total arsenic concentration of 14.4 ± 1.1 mg kg−1 (n = 3, RSD 7.6%).Determination of arsenic compounds
Portions of the freeze-dried powders (0.2 g) were weighed into 15 cm3 polyethylene tubes and were extracted for 14 h with 10 cm3 of methanol by shaking top over bottom at ambient temperature. The resulting mixtures were centrifuged at 1800 rpm. Aliquots (5.00 cm3 of the supernatants) were transferred into polypropylene tubes and the solutions were evaporated to dryness in a centrifugal lyophiliser (Maxi Dry Lyo, Heto-Holten, Allerød, Denmark) at room temperature. The residues were dissolved in water (5.00 cm3), the solutions were filtered through nylon filters (CAMEO 25NS, Osmonics, Minnetonka, USA; pore size 0.22 μm) and the filtrates were analysed by HPLC-ICPMS. The concentrations of the arsenic compounds in the extracts were quantified with calibration curves established with standard solutions of the corresponding compounds. A calibration curve established with As(V) standard solutions was used for quantification of the thio glycerol and thio phosphate sugars. For quality assurance of speciation analyses AB concentration was measured in the certified reference material DORM-2 (17.1 ± 1.0 mg kg−1), and the results were in the certified range (16.4 ± 1.1 mg kg−1).Results and discussion
Total arsenic in the freshwater mussels
Total arsenic concentrations determined in mussels from the Danube are summarized in Table 1. The values obtained, between 3.8 and 12.8 mg As kg−1, are similar to those reported by Koch et al.5 in Margaritifera sp. (3.1 mg As kg−1) and Anadonta sp. (6.7 mg As kg−1) collected from arsenic-rich rivers in Canada, even though the arsenic concentration in the water of the Danube is low. The Mussel Watch Project,10 which monitored arsenic concentrations in marine mussels and oysters from many sites in the USA, reported an overall average total arsenic concentration of 11.1 ± 3.4 mg kg−1. These data suggest that there may not be a big difference between the arsenic concentrations in marine and freshwater bivalves. This is in contrast to other freshwater biota, such as fish and algae, which have much less (sometimes 100-fold less) arsenic than marine species.11Chromatographic conditions and arsenic compounds in the freshwater mussels
Chromatographic conditions applied for the determination of arsenic compounds in the mussel samples are summarized in Table 2. With chromatographic condition I (cation-exchange), it is possible to separate AB, the glycerol sugar, TMAO, AC and TETRA. These five arsenic compounds are well separated from As(III), As(V), MA, DMA and the phosphate, sulfonate, and sulfate sugars, which elute with or close to the solvent front on the Zorbax 300-SCX column.8Fig. 1 demonstrates the structure of arsenic compounds relevant to this work. Only two of the above-mentioned five cationic compounds, namely AB and the glycerol sugar, were present in the mussel extracts in our work. Fig. 2 shows a sample chromatogram recorded under our cation-exchange chromatographic conditions. Using this system it is possible to quantify AB, glycerol sugar, TMAO, AC and TETRA (Table 2). In the extract of this sample only the glycerol arsenosugar could be quantified. | ||
| Fig. 2 Cation-exchange chromatogram of a methanol-extract of Unio pictorum collected at Dunaföldvár (column: ZORBAX 300-SCX, column temperature: 30 °C, mobile phase: 10 mM pyridine at pH 2.6, injection volume: 20 mm3, flow rate: 1.5 cm3 min−1). | ||
As(III), DMA, MA, the phosphate sugar, As(V), the sulfonate and sulfate sugars can be separated on the PRP-X100 anion-exchange column with 20 mM aqueous NH4H2PO4 at pH 5.6 as mobile phase (Chromatographic condition II). Because As(III) elutes at the solvent front, it co-elutes with the cationic arsenicals and hence cannot be reliably quantified under these chromatographic conditions. Only three of the above-mentioned anionic compounds, namely DMA, the phosphate sugar and As(V), were detected in the extracts. An additional strongly retarded arsenical, however, was also present in all samples. This compound (retention time 18.1 min), showed the same chromatographic behavior as one of the thio arsenosugars recently reported in canned marine mussels by Schmeisser et al.13 When the extracts were chromatographed under anion-exchange conditions at pH 10.3 (‘Chromatographic condition III’, Table 2), the compound eluted at a retention time of 5.1 min and a second additional arsenical eluted at 7.9 min (Fig. 3). These two arsenicals were identified as the recently reported thio glycerol arsenosugar and the thio phosphate arsenosugar based on comparison of the retention times with synthetic standards and by spiking experiments (Fig. 3). The retention of these two thio arsenosugars is unusual and quite different from their oxo arsenosugar analogues. Similar unusual chromatographic behavior was noted in the first report of a thio arsenical, namely dimethylarsinothioylacetic acid [(CH3)2As(S)CH2COOH], which was recently identified in sheep urine.14 Possibly, the strong retention of these thio arsenicals can be explained by non-ionic interactions of the As![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) S group with the stationary phase, but this needs further investigation.
S group with the stationary phase, but this needs further investigation.
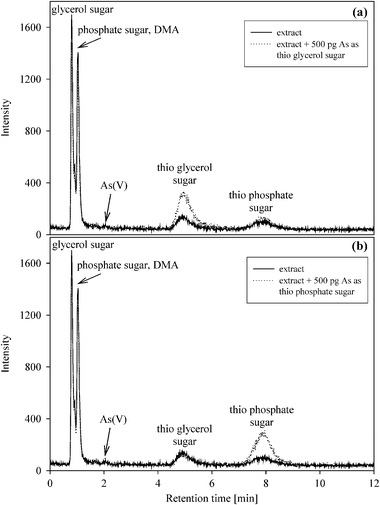 | ||
| Fig. 3 Anion-exchange chromatograms of a methanol-extract of Unio pictorum collected at Dunaföldvár (column: Hamilton PRP-X100, column temperature: 40 °C, mobile phase: 20 mM NH4HCO3 at pH 10.3, injection volume: 20 mm3, flow rate: 1.5 cm3 min−1). | ||
Table 1 shows the concentration of the arsenic species detected by HPLC-ICPMS analysis of the extracts of the mussel samples. We note that the sum of the arsenic species detected represented only 11–22% of the total arsenic in the mussels. In our previous experience in quantification of arsenic species in aquatic animals we have not encountered such large discrepancies. Reasons for these discrepancies will be examined in future studies.
The arsenic distributions in the extracts were similar for the different mussel samples irrespective of their origin. In addition to As(V), six organic arsenic species were found in the extracts: AB, DMA, the glycerol sugar, the phosphate sugar, the thio glycerol sugar and the thio phosphate sugar.
The patterns of the arsenic compounds found in these freshwater mussel samples have several interesting features. First, AB was detected in only three samples and in none of the cases did it exceed 4.4% of the sum of species eluting from the HPLC column. The dominant presence of AB in marine mussels has been well established and AB has also been proposed as the major compound in extracts of the freshwater mussel Dreissena polymorpha by Slejkovec et al.7 In the extract of Margaritifera sp., however, AB was present only in trace amounts and in the case of Anadonta sp. it was not detected at all.5
The second interesting result is that the glycerol sugar and the phosphate sugar are the major identified extractable constituents in the freshwater mussels. On average, these two compounds each constituted about 30% of the summed arsenic species following chromatography. Arsenosugars have already been detected in the marine mussel Mytilus edulis, but as minor compounds.15 Larsen et al.16 reported that arsenosugars can occur as major arsenicals in mussels living in hydrothermal vents 3500 m below the ocean surface as well. Our data confirm the results of Koch et al.,5 who also found mainly arsenosugars in the extracts of two investigated freshwater mussels.
The third point of interest is that all extracts of the freshwater mussels contained two thio sugar species: the thio glycerol sugar and the thio phosphate sugar. These compounds were first reported only recently in marine molluscs.13,17 In the extracts of the freshwater mussels, thio sugar species were measured in the range of 25–195 μg As kg−1 (6–16% of the summed arsenic species) in the case of the thio glycerol sugar and in the range of 86–235 μg As kg−1 (10–22% of the summed arsenic species) in the case of the thio phosphate sugar. It is surprising that these new arsenicals, which appear to be significant species in mussels, have not previously been detected. The fact that the thio sugar compounds elute from the anion-exchange column at long retention times under the commonly used chromatographic conditions could have hampered their observation in previous decades. The observed instability of these compounds (they readily convert in air to the corresponding arsine oxides) may be another reason why this class of compounds has not been found earlier. This is the first work reporting the presence of thio arsenic species in freshwater organisms, and supports the assumption that the thio analogues of different arsenic compounds might be more widespread in the environment.
In general, arsenosugars are the dominant extractable forms of arsenic in the investigated mussel samples constituting 83–95% of the summed arsenic species from HPLC. A higher percentage of the sugars are present as oxo sugars (53–74%) while their thio analogues account for 20–38%. The probable source of the oxo arsenosugars found in the mussel samples is ingested phytoplankton. The origin of the thio-analogues is not known yet, but it should be mentioned that the whole soft body of the mussels was investigated—hence, it is possible that microbiological conversion took place in the gut to give the thio compounds. It cannot be excluded that the long storage time has some influence on the formation of the thio arsenosugars but the observation that thio sugars were also present in fresh mussels from the Danube18 suggests that these compounds are naturally occurring arsenicals. Further investigation of this question is in progress.
The subsequent fate of these arsenicals in the freshwater ecosystem is of interest. They may be taken up by other organisms via feeding or be delivered to the sediments after senescence and decomposition of old mussels. The low to undetectable concentrations of AB in the mussels suggest that the cycle of arsenic is quite different in the freshwater food-web compared to marine systems, even if the feeding habits of the mussels are similar. The significant presence of unknown arsenic, not measurable by the methods of speciation analysis based on HPLC-ICPMS used in this study, suggests unknown toxicological properties of the investigated samples which merits further work.
Acknowledgements
Financial support of this work by the FWF project P16088-N03, the WTZ project A31/2003 (Wissenschaftlich-Technische Zusammenarbeit Österreich Ungarn), and the EC project No. QLK4-CT-2001-00264 is gratefully acknowledged. The authors express their gratitude to Béla Csányi for the sample collection.References
- C. Gundacker, Environ. Pollut., 2000, 110, 61–71 CrossRef CAS.
- J. M. Roper, D. S. Cherry, J. W. Simmers and H. E. Tatem, Environ. Pollut., 1996, 94, 117–129 CrossRef CAS.
- K. A. Francesconi and J. S. Edmonds, Adv. Inorg. Chem., 1997, 44, 147–189 CAS.
- K. A. Francesconi and D. Kuehnelt, in Environmental chemistry of arsenic, ed. W. T. Frankenberger, Jr., Marcel Dekker, New York, 2002, pp. 51–94 Search PubMed.
- I. Koch, K. J. Reimer, A. Beach, W. R. Cullen, A. Gosden and V. W. M. Lai, in Arsenic exposure and health effects IV, ed. W. R. Chappell, C. O. Abernathy and R. L. Calderon, Elsevier, Amsterdam, 2001, pp. 115–123 Search PubMed.
- D. Kuehnelt and W. Goessler, in Organometallic compounds in the environment, ed. P. J. Craig, John Wiley & Sons Ltd., Chichester, 2003, pp. 223–275 Search PubMed.
- Z. Slejkovec, A. R. Byrne, B. Smodis and M. Rossbach, Fresenius’ J. Anal. Chem., 1996, 354, 592–595 CAS.
- W. Goessler and M. Pavkov, Analyst, 2003, 128, 796–802 RSC.
- K. A. Francesconi, J. S. Edmonds, R. V. Stick, B. W. Skelton and A. H. White, J. Chem. Soc., Perkin Trans. 1, 1991, 2707–2716 RSC.
- N. J. Valette-Silver, G. F. Rindel, E. A. Crecelius, H. Windom, R. G. Smith and S. S. Dolvin, Mar. Environ. Res., 1999, 48, 311–333 CrossRef CAS.
- J. Zheng and H. Hintelmann, J. Anal. At. Spectrom., 2004, 19, 191–195 RSC.
- G. Raber, K. A. Francesconi, K. J. Irgolic and W. Goessler, Fresenius’ J. Anal. Chem., 2000, 367, 181–188 CrossRef CAS.
- E. Schmeisser, R. Raml, K. A. Francesconi, D. Kuehnelt, A.-L. Lindberg, C. Sörös and W. Goessler, Chem. Commun., 2004, 1824–1825 RSC.
- H. R. Hansen, R. Pickford, J. Thomas-Oates and J. Feldmann, Angew. Chem., Int. Ed., 2004, 43, 337–340 CrossRef CAS.
- T. Dagnac, A. Padró, R. Rubio and G. Rauret, Talanta, 1999, 48, 763–772 CrossRef CAS.
- E. H. Larsen, C. R. Quétel, R. Munoz, A. Fiala-Medioni and O. F. X. Donard, Mar. Chem., 1997, 57, 341–346 CrossRef CAS.
- M. W. Fricke, P. A. Creed, A. N. Parks, J. A. Shoemaker, C. A. Schwegel and J. T. Creed, J. Anal. At. Spectrom., 2004, 19, 1454–1459 RSC.
- R. Schaffer, C. S. Soeroes, P. Fodor, K. A. Francesconi, W. Goessler, F. Raml, N. Kienzl and D. Kuehnelt, 2005, unpublished data.
| This journal is © The Royal Society of Chemistry 2005 |