Single-strand helical complexes constructed from 2-pyridinyl-3-pyridinylmethanone: tuning the helical pitch length by variation of metal cation and/or counter anion
Xu-Dong
Chen
and
Thomas C. W.
Mak
*
Department of Chemistry, The Chinese University of Hong Kong, Shatin, New Territories, Hong Kong SAR, P. R. China
First published on 27th September 2005
Abstract
The new ligand 2-pyridinyl-3-pyridinylmethanone (L) proves to be an excellent building block for the construction of single-strand helical architectures. A series of helical complexes have been synthesized by the reaction of L with various metal salts, in which L exhibits three kinds of coordination modes involving two kinds of bridging conformations, resulting in four types of single-strand helical chains. The counter anions in the series of 21 helical silver(I) complexes {[Ag(L)]X}∞ (X = NO3, 1; PF6, 2; BF4, 3; ClO4, 4; CF3CO2, 5; CF3SO3, 6) are fully or partially embedded inside the cylindrical helix, and the pitch length corresponds not only to the size of the anion but also to its manner of docking into the groove of the helix. Formation of the helical structure in {[Cu(L)(CH3CN)(H2O)(ClO4)]ClO4}∞ (7) is driven by Ow–H⋯O (perchlorate) hydrogen bonding that leads to a stable triangular motif which rigidly fixes the configuration of the helix. In {[Co(L)(H2O)3](ClO4)2·2H2O}∞ (8) and {[Zn(L)(H2O)3](CF3SO3)2·H2O}∞ (9), similar helical chains without anion embedment suggest that the pitch length can be tuned by the size of metal cations. Notably, complex {[Ag(L)]CF3SO3}∞ (10), a conformational polymorph of 6, has a 41 helix induced by argentophilic interaction.
Introduction
The helical organization of molecules is a commonly observed phenomenon throughout nature, and hence a comprehensive understanding of the factors governing the formation of helical structures is of great importance not only in mimicking nature but also in preprogramming molecules to give specific architecture and defined functionality.1–5 The construction of metal–organic helical coordination polymeric structures has been achieved with strand-like ligands that undergo wrapping to generate helicity, and short bridging ligands with helical information encoded by conformational restriction mostly lead to single-strand helices.4,6,7The self-organization process of ligands with metal ions to form defined architectures and functionalities depends on the selection of optimal components such as the coordination geometry of the metal ions, the binding site of the donating atoms, and the length of the spacers that link the binding sites.2–4,8,9 Since ligands generally have specific backbones and linking information and the coordination geometries of various metal ions are so different, it remains a challenge to develop useful ligands for the generation of helical moieties adaptive to a wide range of metal cations and counter anions.10–12
In contrast to extensive studies on the coordination chemistry of di-2-pyridinylmethanone (di-2-pyridyl ketone),13 metal complexes of its N-positional isomer 2-pyridinyl-3-pyridinylmethanone (L) has remained unexplored so far (Scheme 1). With its two differentiable pyridyl rings bearing variable dihedral angles within a limited range, this flexible ligand is potentially capable of linking metal ions into an infinite helix. Here we report a series of helical complexes generated from L and various metal ions (Ag+, Cu2+, Co2+ and Zn2+) and counter anions (NO3−, PF6−, BF4−, ClO4−, CF3SO3− and CF3CO2−).
 | ||
| Scheme 1 | ||
Experimental
General
Diethyl ether was purchased from LAB-SCAN and further refluxed over sodium and benzophenone. All other chemicals were obtained commercially from Aldrich and used without further purification.Elemental analyses of C, H and N were performed by the MEDAC LTD Brunel Science Centre, United Kingdom. IR spectra were recorded with a Nicolet Impact 420 FT-IR spectrometer using KBr pellets. 1H NMR spectra were taken at 300 Hz with a Bruker-300 spectrometer using CDCl3 as solvent. Mass spectrometry was conducted on a ThermoFinnigan MAT 95 XL spectrometer.
Syntheses
The synthesis and characterization of conformational polymorphs 6 (space group P21/c) and 10 (space group P41212 and P43212) with the same structural formula {[Ag(L)]CF3SO3}∞ have been reported in a recent communication.14
X-Ray diffraction studies
Single-crystal X-ray diffraction measurements were carried out on a Bruker SMART 1000 CCD diffractometer operating at 50 KV and 30 mA using Mo-Kα radiation (λ = 0.71073 Å). Each selected crystal was mounted inside a Lindemann glass capillary for data collection at 293 K. Data collection and reduction were performed using the SMART and SAINT software.15 An empirical absorption correction was applied using the SADABS program.16 All the structures were solved by direct methods and refined by full-matrix least squares on F2 using the SHELXTL program package.17 All non-hydrogen atoms were subjected to aniostropic refinement, and all hydrogen atoms except those of water molecules were added in idealized positions and refined isotropically. Hydrogen atoms of aqua ligands in complexes 7–9 and hydrogen atoms of the lattice water molecules in complex 9 were located from difference electronic Fourier maps. Hydrogen atoms of the lattice water molecules in complex 9 could not be located. The corresponding counter anions in complexes 1–5 and 7–8 were treated using a disordered model. Crystal data and details of the refinement for 1–10 are summarized in Table 1.CCDC reference numbers 278821–278828.
See http://dx.doi.org/10.1039/b510569b for crystallographic data in CIF or other electronic format.
| Complex | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 |
|---|---|---|---|---|---|---|---|---|---|---|
| a R 1 = Σ‖Fo| − |Fc‖/Σ|Fo|. b wR 2 = {Σ[w(Fo2 − Fc2)2]/Σ[w(Fo2)2]}1/2. | ||||||||||
| Empirical formula | C11H8N3O4Ag | C11H8F6N2OPAg | C11H8BF4N2OAg | C11H8ClN2O5Ag | C13H8F3N2O3Ag | C12H8F3N2O4SAg | C13H13Cl2N3O10Cu | C11H18Cl2N2O14Co | C13H16F6N2O11S2Zn | C12H8F3N2O4SAg |
| M | 354.07 | 437.03 | 378.87 | 391.51 | 405.08 | 441.13 | 505.70 | 532.10 | 619.77 | 441.13 |
| Crystal size/mm | 0.46 × 0.36 × 0.25 | 0.20 × 0.18 × 0.09 | 0.45 × 0.17 × 0.16 | 0.43 × 0.24 × 0.17 | 0.45 × 0.32 × 0.30 | 0.45 × 0.23 × 0.15 | 0.43 × 0.43 × 0.38 | 0.34 × 0.25 × 0.14 | 0.50 × 0.42 × 0.24 | 0.34 × 0.30 × 0.17 |
| Crystal system | Monoclinic | Monoclinic | Monoclinic | Monoclinic | Monoclinic | Monoclinic | Orthorhombic | Monoclinic | Monoclinic | Tetragonal |
| Space group | P21/c | P21/c | P21/c | P21/c | P21/c | P21/c | Pbca | P21/n | P21/c | P41212 |
| a/Å | 7.9514(6) | 8.6733(7) | 8.0172(5) | 8.1126(9) | 7.7407(5) | 9.5431(6) | 18.919(1) | 13.0805(8) | 10.1623(5) | 10.2355(4) |
| b/Å | 12.1163(9) | 12.193(1) | 12.4067(8) | 12.513(1) | 13.6362(8) | 12.4438(8) | 8.1263(5) | 11.1580(7) | 11.5785(5) | 10.2355(4) |
| c/Å | 12.4823(9) | 13.270(1) | 13.2083(9) | 13.418(2) | 13.5921(9) | 13.3198(9) | 24.802(1) | 13.8114(8) | 19.8500(9) | 28.105(2) |
| α/° | 90 | 90 | 90 | 90 | 90 | 90 | 90 | 90 | 90 | 90 |
| β/° | 107.077(1) | 101.673(2) | 110.355(1) | 111.047(2) | 109.424(1) | 105.165(1) | 90 | 95.398(1) | 97.017(1) | 90 |
| γ/° | 90 | 90 | 90 | 90 | 90 | 90 | 90 | 90 | 90 | 90 |
| V/Å3 | 1149.5(2) | 1374.4(2) | 1231.8(1) | 1271.2(3) | 1353.0(2) | 1526.7(2) | 3813.0(4) | 2006.9(2) | 2318.1(2) | 2944.4(3) |
| Z | 4 | 4 | 4 | 4 | 4 | 4 | 8 | 4 | 4 | 8 |
| D calc/g cm−3 | 2.046 | 2.112 | 2.043 | 2.046 | 1.989 | 1.919 | 1.762 | 1.761 | 1.776 | 1.990 |
| μ(Mo-Kα)/mm−1 | 1.768 | 1.654 | 1.681 | 1.816 | 1.539 | 1.509 | 1.485 | 1.195 | 1.344 | 1.565 |
| F(000) | 696 | 848 | 736 | 768 | 792 | 864 | 2040 | 1084 | 1248 | 1728 |
| Reflections collected | 4920 | 7267 | 6505 | 6706 | 7177 | 10599 | 19358 | 10620 | 12107 | 15946 |
| Independent reflections (Rint) | 2035 (0.0243) | 2422 (0.0510) | 2165 (0.0327) | 2241 (0.0367) | 2388 (0.0284) | 3927 (0.0338) | 3354 (0.0393) | 3535 (0.0372) | 4087 (0.0350) | 2596 (0.0193) |
| Observed reflections [I > 2σ(I)] | 1791 | 1895 | 1909 | 1834 | 1974 | 2687 | 2742 | 2846 | 3625 | 2490 |
| Parameters | 199 | 226 | 217 | 217 | 226 | 208 | 318 | 340 | 324 | 208 |
| Goodness-of-fit | 1.136 | 1.119 | 1.066 | 1.206 | 1.066 | 1.049 | 1.169 | 1.131 | 1.120 | 1.080 |
| R 1 [I > 2σ(I)]a | 0.0368 | 0.0525 | 0.0365 | 0.0573 | 0.0350 | 0.0536 | 0.0466 | 0.0502 | 0.0460 | 0.0229 |
| wR 2 (all data)b | 0.0966 | 0.1399 | 0.0988 | 0.1526 | 0.0934 | 0.1669 | 0.1235 | 0.1438 | 0.1322 | 0.0582 |
Results and discussion
X-Ray crystallographic characterization showed that complexes 1–9 exhibit a single-strand helical chain structure winding around a 21 axis whereas 10 has a 41 axis.14 The key tectonic element that leads to the wide adaptation of L to a wide variety of metal ions and counter anions in forming helical architectures can be ascribed to its flexible conformation. Rotation of the 2- and 3-pyridyl rings about the respective C–C(carbonyl) single bonds result in a wide range of bridging conformations (angular parameters θ and ϕ, calculated from the torsion angle M–N1⋯N2–M′) corresponding to the size variation of both the metal ions and the counter anions (Fig. 1). Ligand L exhibits two kinds of conformation in this series of helical complexes: in conformation A, the two pyridyl nitrogen atoms are anti-related to generate an obtuse bridging angle θ, while conformation B has a syn-related pair to form an acute bridging angle ϕ. Conformation A is more frequently observed in the metal complexes of L, in which variation of the bridging angle θ yields spring-like helices of different pitch lengths. Compared to the linear coordination geometry of the silver(I) ion,18 the metal(II) ions Cu2+, Co2+ and Zn2+ are each trans-coordinated by the nitrogen atoms of two independent ligands L, its octahedral coordination sphere being completed by the carbonyl oxygen and supplementary ligands such as aqua ligand and acetonitrile molecule. Selected bond lengths and angles, together with selected shape parameters of complexes 1–10 are listed in Table 2.|
|
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Complex | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 |
| M–N bond lengths/Å | 2.177(3) | 2.160(5) | 2.151(4) | 2.162(6) | 2.212(3) | 2.163(4) | 1.997(4) | 2.121(4) | 2.139(3) | 2.170(3) |
| 2.189(3) | 2.170(5) | 2.175(4) | 2.179(6) | 2.242(3) | 2.176(4) | 1.997(4) | 2.123(4) | 2.141(3) | 2.194(3) | |
| N1–M–N2′ angle/° | 172.5(1) | 177.7(2) | 175.9(1) | 174.8(2) | 167.4(1) | 176.4(2) | 167.5(2) | 166.4(1) | 162.7(1) | 157.9(1) |
| M⋯O (carbonylic) distance/Å | 2.838(4) | 2.697(5) | 2.692(3) | 2.720(6) | 2.698(3) | 2.757(4) | 2.008(3) | 2.127(3) | 2.244(2) | 2.744(3) |
| C5–C6–C8 angle/° | 118.5(3) | 119.0(5) | 119.1(4) | 119.9(6) | 121.0(3) | 118.1(4) | 124.4(4) | 123.8(4) | 122.1(3) | 119.5(3) |
| N1–C5–C6–O torsion angle/° | 31.1 | 26.9 | 27.6 | 26.9 | −26.2 | −24.1 | 2.2 | −3.2 | −9.7 | 23.2 |
| O–C6–C8–C7 torsion angle/° | −140.2 | −134.2 | −140.0 | −140.5 | 142.9 | 136.0 | 38.6 | 140.6 | 141.9 | −133.1 |
| Dihedral angle of the two pyridyl rings/° | 65.2 | 68.2 | 64.2 | 63.8 | 57.3 | 65.8 | 44.5 | 40.9 | 44.1 | 69.3 |
| M–N1⋯N2–M′ torsion angle (θ or ϕ)/° | −109.2 | −111.5 | 113.3 | 113.0 | 119.4 | −111.1 | 43.9 | 146.1 | 145.9 | −95.7 |
| Pitch length of helix/Å | 12.116 | 12.193 | 12.407 | 12.513 | 13.636 | 12.444 | 8.126 | 11.158 | 11.578 | 28.105 |

 | ||
| Fig. 1 Two kinds of bridging conformations of 2-pyridinyl-3-pyridinylmethanone (L) in its coordination complexes, leading to infinite helices with different helical pitch lengths. | ||
Reaction of L with various silver(I) salts gives rise to complexes 1–6 formulated as {[Ag(L)]X}∞ (X = NO3, 1; PF6, 2; BF4, 3; ClO4, 4; CF3CO2, 5; CF3SO3, 6). All six complexes comprise an infinite single-strand helix constituted from the alternate linkage of silver(I) cations and ligands L, and each helical pitch contains two silver(I)–L units (Fig. 2). The silver(I) atom adopts an almost linear coordination geometry while L acts as a bidentate bridging ligand with its carbonyl oxygen atom weakly interacting with the silver(I) atom at a distance ranging from 2.69 Å to 2.84 Å. The cationic helix is charge-balanced by two parallel columns of the corresponding counter anions that are pinched fully or partially into the grooves. The right-handed and left-handed helixes co-exist in each silver(I) complex, and they are arrayed in an alternate fashion to generate a racemic crystal.
 | ||
| Fig. 2 Part (a) to (f) show the infinite helical chains with imbedded counter anions in complexes 1–6, respectively. The disordered atoms in the anions are omitted for clarity. Color scheme: C, light gray; H, white; O, red; N, deep blue; Ag, purple; Cl, light green; F, green; S, yellow; P, deep green; B, orange. | ||
Comparison of the crystal structures of complexes 1–6 indicates that the helical pitch lengths can be correlated with the size of the embedded counter anions (Fig. 2).19 In another words, the pitch length of the spring-like helix can be tuned by the incorporation of counter anions with different sizes. With NO3− as the smallest anion within this series, complex 1 exhibits a helical pitch length of 12.116 Å, being significantly shorter than that of complex 5 (13.636 Å) with the larger CF3CO2− anion.19 The PF6−, BF4− and ClO4− anions of intermediate sizes lead to similar medium pitch lengths at 12.193, 12.407 and 12.513 Å for 2, 3 and 4, respectively. It is notable that, with CF3SO3− as the largest within this series of anions, complex 6 only exhibits a helical pitch length of 12.444 Å. This apparent anomaly can be attributed to the fact that the CF3SO3− anion is only partially mounted in the helix with the CF3 group exposed (Fig. 2f). Hence the “effective volume” of the CF3SO3− anion incorporated in the helix is similar to those of the PF6−, BF4− and ClO4− anions. Therefore, the pitch length of a spring-like {[Ag(L)]X}∞ 21 helical chain is dominated not only by the size of the inlaid counter anion but also by its mode of fitting to the grooves inside the helix.
This self-adjustment of pitch length of the helix is realized mainly by adaptive variation of the bridging angle (θ) in ligand L. For complex 1, the value of θ is −109.2° for the smallest NO3− anion in this series, while in complex 5 the corresponding value is 119.4° for the CF3CO2− anion with the largest incorporated volume. Taking into account the partial insertion of CF3SO3− into the groove of the helix, the value of θ is similar for complexes 2, 3, 4 and 6, being −111.5, 113.3, 113.0 and −111.1°, respectively. The full incorporation of the larger CF3CO2− anion into the helix leads to significant structural differences of complex 5 compared with complexes 1–4 and 6 (Table 2). The angle between the two single bonds about the carbonyl group is 121.0° for 5, bending in the opposite manner as compared to the value of 118.1–119.9° for the other five complexes. Furthermore, the dihedral angle between the pyridyl rings of ligand L in complex 5 (57.3°) is much smaller than those of the others (in the range 63.8–68.2°), being consistent with its longer pitch length. Concerning the coordination environment of the silver(I) atom, the Ag–N bond length is significantly longer for complex 5 (2.212 and 2.242 Å) as compared with those in other complexes (ranging from 2.151 to 2.189 Å), and the N–Ag–N angle deviates from linearity more for 5 (167.4°) than those in other complexes (172.5 to 177.7°).
Though complexes 1–6 all crystallized in space group P21/c, they exhibit two kinds of packing modes in their crystal structures: mode I for 1, 2 and 6; and mode II for 3–5, as shown in Fig. 3. Both modes comprise layers that are assembled from infinite single-strand helices by weak interaction between the silver(I) atom and the counter anion (ranging from 2.7 to 3.6 Å), and such layers are further linked into a three-dimensional network by C–H⋯anion interactions. The nature of the counter anion obviously plays an important role in dictating the packing mode that is preferentially adopted in a silver(I) complex, but the interplay of various weak intermolecular interactions makes it difficult to draw reliable predictions for specific anions.
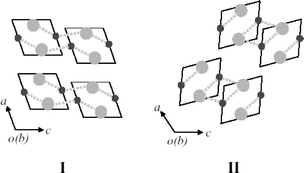 | ||
| Fig. 3 Two kinds of packing modes in complexes 1–6: mode I for 1, 2 and 6; and mode II for 3–5. Each parallelogram represents the projection of a single helix. The small circles in dark gray and large circles in light gray represent silver atoms and anions, respectively. The dotted lines indicate weak interactions between the silver cations and anions. | ||
In complex 7, ligand L bridges consecutive copper(II) atoms in syn mode B, as opposed to the anti mode A observed in complexes 1–6 (Fig. 1), yielding a different infinite single-strand helical chain. In this chain, the slightly-distorted pyramidal coordination requirement of the copper(II) atom is satisfied by two consecutive L ligands acting in chelating and monodentate modes, an acetonitrile molecule and an aqua ligand (Fig. 4). At the opposite side of the pyramidal apex, an oxygen atom from a perchlorate group has a weak interaction with the copper(II) center at a distance of 2.599(4) Å. Hence the coordination geometry of the copper(II) center can also be seen as distorted octahedral. A similar helical structure has been reported by Feringa et al.,6a in which each copper(I) atom is coordinated by two pyridine N atoms and two imine N atoms in a distorted tetrahedral geometry. It is noteworthy that intramolecular aqua–perchlorate hydrogen bonding (O8A⋯O1w 2.849 Å, O8A⋯H1wA 2.048 Å and O1w–H1wA⋯O8A 165.3°) rigidly fixes the helical structure (calculated for the disordered oxygen atom of the weakly ligated perchlorate anion, which is farthest away from the aqua ligand), as illustrated in Fig. 4(b).20Via these hydrogen bonds, the crystallographically equivalent perchlorate anions are pinched inside the cylindrical helix in two columns. The second type of perchlorate anion is arranged in external columns that charge-balance the cationic helix. The pitch length of the helix in complex 7 is significantly shorter at 8.126 Å due to the specific ligation conformation adopted by L. The P- and M-helixes are packed alternately and cross-linked by C–H⋯O(perchlorate) hydrogen bonds, so that 7 exists as a racemate in the bulk material.
 | ||
| Fig. 4 (a) Infinite cationic helical chain in complex 7. (b) Helical structure with triangular motif consolidated by Ow–H⋯O(perchlorate) hydrogen bonding in 7. Color scheme: C, light gray; H, white; O, red; N, deep blue; Cu, turquoise; Cl, green. | ||
In complexes 8 and 9, L exhibits a similar anti linking mode A as that in complexes 1–6, in contrast to the syn mode in 7 (Fig. 1), bridging the cobalt(II) or zinc(II) atoms into an infinite cationic single-strand helical chain (Fig. 5). The helices in 8 and 9 are very similar: besides the three binding sites from a pair of L ligands, both complexes have their octahedral metal coordination sphere completed by a facial group of the aqua ligands. The P- and M-helical chains are cross-linked alternately to form a racemic crystal via C–H⋯O(perchlorate) hydrogen bonds and inter-chain hydrogen bonds involving these aqua ligands. The counter anions and lattice water molecules lie outside the helical chains in 8 and 9, leading to the pitch lengths of 11.158 Å and 11.578 Å, respectively. With no pinched anion inside the helix, the pitch length is mainly dominated by the size of the metal cation, as evidenced by the shorter length for the cobalt(II) helix in 8 compared to the zinc(II) helix in 9 (Table 2). This is also supported by the significantly longer M(II)–N bond lengths for 8 (2.121(4) and 2.123(4) Å) than those for 9 (2.139(3) and 2.141(3) Å), as well as a slightly larger value of θ in 8 (146.1°) than that in 9 (145.9°).
 | ||
| Fig. 5 Part (a) and (b) show the cationic infinite helical chains in complexes 8 and 9, respectively. Color scheme: C, light gray; H, white; O, red; N, deep blue; Co, turquoise; Zn, green. | ||
Three kinds of coordination mode of L are observed in a series of helical complexes, namely bidentate anti-bridging (in 1–6, and 10), tridentate syn-bridging (in 7) and tridentate anti-bridging (in 8 and 9) (Fig. 6). These coordination modes involve two kinds of bridging conformation A and B that lead to four types of infinite helical chains which can be easily recognized from views along the helical axis (Fig. 7). The cross-section of the infinite helix in complexes 1–6 and 8–9 are rectangular and that in complex 7 is rhombic, while that in complex 10 is square. Different bridging modes of L, as described by the obtuse bridging angle θ in conformation A as compared with the acute bridging angle ϕ in conformation B, account for the fact that the helical pitch lengths in complexes 1–6 and 8, 9 (11.158–13.636 Å) are correspondingly much longer than that in complex 7 (8.126 Å). The longer pitch lengths of complexes 1–6 (12.116–13.636 Å) compared to those of complexes 8 and 9 (11.158 Å and 11.578 Å) can be rationalized on the basis of both the larger cation size and the anion embedment in complexes 1–6.
 | ||
| Fig. 6 Coordination modes of 2-pyridinyl-3-pyridinylmethanone (L). | ||
 | ||
| Fig. 7 Perspective view of the helix along the helical axis: (a) for complexes 1–6; (b) for complex 7; (c) for complexes 8 and 9; and (d) for complex 10. | ||
Studies on the IR spectra of this series of helical complexes revealed that the vibrations of ligand L were affected only to a minor extent in its coordination complexes. As to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching mode, L exhibits a sharp and strong peak at 1668 cm−1 while all its silver(I) complexes show the same peak at 1666 cm−1 except for the value of 1670 cm−1 in complex 5, and its metal(II) complexes 7–9 show this peak at 1662, 1672 and 1672 cm−1, respectively.
O stretching mode, L exhibits a sharp and strong peak at 1668 cm−1 while all its silver(I) complexes show the same peak at 1666 cm−1 except for the value of 1670 cm−1 in complex 5, and its metal(II) complexes 7–9 show this peak at 1662, 1672 and 1672 cm−1, respectively.
Conclusion
The present study has demonstrated that 2-pyridinyl-3-pyridinylmethanone (L) is a versatile building block for the generation of single-strand helical structures with metal cations and counter anions of different sizes under mild conditions. In the series of silver(I) helical complexes 1–6, with the counter anions pinched inside the 21 helix, the pitch lengths correlate with the “effective” anion volume embedded into the grooves. In the cases of complexes 7 and 10, ligand L takes a particular bridging mode in forming specific helical architectures. In complexes 8 and 9 that comprise helical chains with no embedded anion, the pitch lengths are dictated by the size of the metal cations. The present strategy of tuning the helical pitch length by variation of counter anions and even metal cations may be of value in the development of synthetic methodologies in crystal engineering and materials science.Acknowledgements
This work is financially supported by the Hong Kong Research Grants Council (Ref. No. CUHK 402003).References
- M. J. Kelso, R. L. Beyer, H. N. Hoangt, A. S. Lakdawala, J. P. Snyder, W. V. Oliver, T. A. Robertson, T. G. Appleton and D. P. Fairlie, J. Am. Chem. Soc., 2004, 126, 4828–4842 CrossRef CAS.
- (a) C. Piguet, M. Borkovec, J. Hamacek and K. Zeckert, Coord. Chem. Rev., 2005, 249, 705–726 CrossRef CAS; (b) M. Albrecht, Chem. Rev., 2001, 101, 3457–3497 CrossRef CAS; (c) C. Piguet, G. Bernardinelli and G. Hopfgartner, Chem. Rev., 1997, 97, 2005–2062 CrossRef CAS.
- (a) J.-M. Lehn, Angew. Chem., Int. Ed. Engl., 1990, 29, 1304–1319 CrossRef; (b) J. S. Moore, Curr. Opin. Solid State Mater. Sci., 1996, 1, 777–787 CrossRef CAS; (c) D. S. Lawrence, T. Jiang and M. Levett, Chem. Rev., 1995, 95, 2229–2260 CrossRef CAS; (d) D. B. Amabilino and J. F. Stoddart, Chem. Rev., 1995, 95, 2725–2828 CrossRef CAS; (e) H. Yin, G. I. Lee, H. S. Park, G. A. Payne, J. M. Rodriguez, S. M. Sebti and A. D. Hamilton, Angew. Chem., Int. Ed., 2005, 44, 2704–2707 CrossRef CAS; (f) S. Aravinda, S. Datta, N. Shamala and P. Balaram, Angew. Chem., Int. Ed., 2004, 43, 6728–6731 CrossRef CAS.
- A. E. Rowan and R. J. M. Nolte, Angew. Chem., Int. Ed., 1998, 37, 63–68 CrossRef CAS.
- M. J. Hannon and L. J. Childs, Supramol. Chem., 2004, 16, 7–22.
- (a) O. J. Gelling, F. van Bolhuis and B. L. Feringa, J. Chem. Soc., Chem. Commun., 1991, 917–919 RSC; (b) J.-R. Li, X.-H. Bu, J. Jiao, W.-P. Du, X.-H. Xu and R.-H. Zhang, Dalton Trans., 2005, 464–474 RSC; (c) P. K. Bowyer, K. A. Porter, A. D. Rae, A. C. Willis and S. B. Wild, Chem. Commun., 1998, 1153–1154 RSC.
- (a) Y.-T. Wang, M.-L. Tong, H.-H. Fan, H.-Z. Wang and X.-M. Chen, Dalton Trans., 2005, 424–426 RSC; (b) K. Biradha, C. Seward and M. J. Zaworotko, Angew. Chem., Int. Ed., 1999, 38, 492–495 CrossRef CAS; (c) U. Siemeling, I. Scheppelmann, B. Neumann, A. Stammler, H.-G. Stammler and J. Frelek, Chem. Commun., 2003, 2236–2237 RSC; (d) F. M. Tabellion, S. R. Seidel, A. M. Arif and P. J. Stang, Angew. Chem., Int. Ed., 2001, 40, 1529–1532 CrossRef CAS; (e) T. Ezuhara, K. Endo and Y. Aoyama, J. Am. Chem. Soc., 1999, 121, 3279–3283 CrossRef CAS; (f) M. A. Withersby, A. J. Blake, N. R. Champness, P. Hubberstey, W.-S. Li and M. Schröder, Angew. Chem., Int. Ed. Engl., 1997, 36, 2327–2329 CrossRef CAS; (g) E.-Q. Gao, S.-Q. Bai, Z.-M. Wang and C.-H. Yan, J. Am. Chem. Soc., 2003, 125, 4984–4985 CrossRef CAS; (h) Y.-T. Fu, V. M. Lynch and R. J. Lagow, Chem. Commun., 2004, 1068–1069 RSC.
- J.-M. Lehn, Supramolecular Chemistry-Concepts and Perspectives, VCH, Weinheim, 1995 Search PubMed.
- (a) O.-S. Jung and C. G. Pierpont, J. Am. Chem. Soc., 1994, 116, 2229–2230 CrossRef CAS; (b) D. Venkarataraman, G. B. Gardner, S. Lee and J. S. Moore, J. Am. Chem. Soc., 1995, 117, 11600–11601 CrossRef CAS; (c) O. M. Yaghi, H. Li, C. Davis, D. Richardson and T. L. Groy, Acc. Chem. Res., 1998, 31, 474–484 CrossRef CAS; (d) H. Gudbjartson, K. Biradha, K. M. Poirier and M. J. Zaworotko, J. Am. Chem. Soc., 1999, 121, 2599–2600 CrossRef CAS; (e) S. S.-Y. Chui, S. M.-F. Lo, J. P. H. Charmant, A. G. Orpen and I. D. Williams, Science, 1999, 283, 1148–1150 CrossRef CAS.
- V. Berl, I. Huc, R. G. Khoury, M. J. Krische and J.-M. Lehn, Nature (London), 2000, 407, 720–723 CrossRef CAS.
- (a) S. Hanessian, A. Gomtsyan, M. Simard and S. Roelens, J. Am. Chem. Soc., 1994, 116, 4495–4496 CrossRef CAS; (b) S. Hanessian, M. Simard and S. Roelens, J. Am. Chem. Soc., 1995, 117, 7630–7645 CrossRef CAS; (c) S. G. Telfer, T. Sato and R. Kuroda, Angew. Chem., Int. Ed., 2004, 43, 581–584 CrossRef CAS.
- (a) J.-M. Lehn, Chem. Eur. J., 2000, 6, 2097–2102 CrossRef CAS; (b) I. E. Rowan and R. J. M. Nolte, Angew. Chem., Int. Ed., 1998, 37, 62–68; (c) M. C. Feiters and R. J. M. Nolte, Adv. Supramol. Chem., 2000, 6, 41–156 Search PubMed.
- (a) K. N. Crowder, S. J. Garcia, R. L. Burr, J. M. North, M. H. Wilson, B. L. Conley, P. E. Fanwick, P. S. White, K. D. Sienerth and R. M. Granger, Inorg. Chem., 2004, 43, 72–78 CrossRef CAS and references therein; (b) G. S. Papaefstathiou and S. P. Perlepes, Comments Inorg. Chem., 2002, 23, 249–274 CrossRef CAS and references therein.
- X.-D. Chen and T. C. W. Mak, Chem. Commun., 2005, 4417–4419 RSC.
- Bruker, SMART 5.0 and SAINT 4.0 for Windows NT, Area Detector Control and Integration Software, Burker Analytical X-Ray Systems Inc., Madison, WI, 1998 Search PubMed.
- G. M. Sheldrick, SADABS: Program for Empirical Absorption Correction of Area Detector Data, University of Göttingen: Göttingen, Germany, 1996 Search PubMed.
- G. M. Sheldrick, SHELXTL 5.1 for Windows NT: Structure Determination Software Programs, Bruker Analytical X-ray Systems, Inc., Madison, WI, 1997 Search PubMed.
- L. C. Carlucci, G. Ciani, D. W. V. Gudenberg and D. M. Proserpio, Inorg. Chem., 1997, 36, 3812–3813 CrossRef CAS.
- O.-S. Jung, Y. J. Kim, Y.-A. Lee, J. K. Park and H. K. Chae, J. Am. Chem. Soc., 2000, 122, 9921–9925 CrossRef CAS.
- K. Biradha, CrystEngComm, 2003, 5, 374–384 RSC.
| This journal is © The Royal Society of Chemistry 2005 |