Preparation of chalcogenolato-bridged dinuclear Tp*Rh–Cp*Ru complexes (Tp* = hydrotris(3,5-dimethylpyrazol-1-yl)borate, Cp* = η5-pentamethylcyclopentadienyl) and binding of dioxygen to their Ru sites
Shoken
Nagao
a,
Hidetake
Seino
a,
Masanobu
Hidai
b and
Yasushi
Mizobe
*a
aInstitute of Industrial Science, The University of Tokyo, Komaba, Meguro-ku, Tokyo 153-8505, Japan. E-mail: ymizobe@iis.u-tokyo.ac.jp
bDepartment of Materials Science and Technology, Faculty of Industrial Science and Technology, Tokyo University of Science, Noda, Chiba 278-8510, Japan
First published on 12th August 2005
Abstract
Reactions of [Tp*Rh(coe)(MeCN)] (1; Tp* = hydrotris(3,5-dimethylpyrazol-1-yl); coe = cyclooctene) with one equiv of diphenyl dichalcogenides PhEEPh (E = Se, Te) afforded the mononuclear RhIII complexes [Tp*Rh(EPh)2(MeCN)] (2b: E = Se; 2c: E = Te), as reported previously for the formation of [Tp*Rh(SPh)2(MeCN)] (2a) from the reaction of 1 and PhSSPh. Complexes 2a–2c were treated with the RuII complex [(Cp*Ru)4(µ3-Cl)4] (Cp* = η5-C5Me5) in THF at room temperature, yielding the chalcogenolato-bridged dinuclear complexes [Tp*RhCl(µ-EPh)2RuCp*(MeCN)] (3). Complex 3a (E = S) in solution was converted slowly into a mixture of 3a and the sterically less encumbered dinuclear complex [Tp*RhCl(SPh)(µ-η1-S-η6-Ph)RuCp*] (4a) at room temperature. In 4a, one SPh group binds only to the Rh center as a terminal ligand, while the other SPh group bridges the Rh and Ru atoms by coordinating to the former at the S atom and to the latter with the Ph group in a π fashion. The Se analogue 3b also underwent a similar transformation under more forcing conditions, e.g. in benzene at reflux, whereas formation of the µ–η1-Te-η6-Ph complex was not observed for the Te analogue 3c even under these forcing conditions. When complexes 3 was dissolved in THF exposed to air, the MeCN ligand bound to Ru was substituted by dioxygen to give the peroxo complexes [Tp*RhCl(µ-EPh)2RuCp*(η2-O2)] (5a: E = S; 5b: E = Se; 5c: E = Te). X-Ray analyses have been undertaken to determine the detailed structures for 2c, 3a, 3b, 4a, 5a, 5b, and 5c.
Introduction
Complexes containing the hydrotris(pyrazol-1-yl)borate or its congeners with the substituted pyrazole rings as the coligand are currently attracting significant attention.1 In a previous paper,2 we reported that the reactions of Rh pyrazolylborate complexes [Tp′Rh(coe)(MeCN)] (Tp′ = Tp* (1), Tp; Tp* = hydrotris(3,5-dimethylpyrazol-1-yl); Tp = hydrotris(pyrazol-1-yl); coe = cyclooctene) with a range of organic disulfides RSSR gave a new class of mononuclear and dinuclear Rh thiolato complexes containing Tp′ as the ancillary ligand, and from the mononuclear bis(benzenethiolato) complex [Tp*Rh(SPh)2(MeCN)] (2a), non-symmetrical dinuclear complexes with two bridging SPh ligands [Tp*RhCl(µ-SPh)2MCp*Cl] (M = Rh, Ir; Cp* = η5-C5Me5) can be derived by the subsequent reactions with [(Cp*MCl)2(µ-Cl)2]. Homo- and heterometallic dinuclear complexes obtained in this previous study represent the first chalcogenolato-bridged dinuclear complexes containing Tp′Rh units.Now we have extended the latter reactions to that with the RuII complex [(Cp*RuCl)4(µ3-Cl)4] and found that the analogous bimetallic complex [Tp*RhCl(µ-SPh)2RuCp*(MeCN)] (3a) can be obtained. Interestingly, 3a in solution has proved to be converted into the singly bridged complex [Tp*RhCl(SPh)(µ–η1-S-η6-Ph)RuCp*] (4a), while exposure of 3a in solution to air affords a dioxygen complex [Tp*RhCl(µ-SPh)2RuCp*(η2-O2)] (5a). In this paper, we wish to describe the details of 3a–5a together with the SePh and TePh analogues of 2a–5a derived similarly from 1. It is noteworthy that the selenolato and tellurolato-bridged dinuclear complexes are still limited3 and those having the Tp′Rh moiety are yet unknown.
Results and discussion
Reactions of 1 with PhEEPh (E = Se, Te)
The RhI complex having the Tp* ligand 1 reacted readily with one equiv of PhEEPh (E = Se, Te) in THF to give mononuclear RhIII bis(chalcogenolato) complexes [Tp*Rh(ER)2(MeCN)] (2b: E = Se, 2c: E = Te) (eqn (1)), as observed previously in the reaction of 1 with PhSSPh forming 2a. The reaction with PhSeSePh proceeds more smoothly than that with PhSSPh: the 1H NMR spectra of the reaction mixture indicated that the former reaction terminated within 1 h at room temperature, whereas the latter reaction required ca. 24 h until completion under similar conditions. On the other hand, the reaction with PhTeTePh also takes place quite rapidly at room temperature but does not proceed cleanly. To avoid the formation of intractable by-products, preparation of 2c was undertaken at −20 °C. | (1) |
The 1H NMR data for 2b and 2c are in good agreement with those of 2a, indicating that these new complexes are isomorphous with the thiolato complex 2a as well as its crystallographically characterized congener [Tp*Rh(STol)2(MeCN)] (2a′; Tol = p-MeC6H4). Thus, the spectra of 2b and 2c show the presence of the Tp*, EPh, and MeCN ligands in a ratio of 1 : 2 : 1, where the signals due to the 3,5-dimethylpyrazolyl groups are recorded as two sets with an intensity ratio of 2 : 1, while those ascribable to the Ph groups suggest the equivalence of the two SPh groups in solution. These features were confirmed by the X-ray analysis of 2c, whose ORTEP drawing is shown in Fig. 1. Pertinent bond distances and angles in 2c are listed in Table 1, which correspond well to those in 2a′ except for the elongation of the Rh–Te bonds by ca. 0.3 Å from the Rh–S bonds and the concomitant decrease in the E–Rh–E angle from 94.4° for E = S to 87.1° for E = Te. The difference in the Rh–E bond lengths agrees well to that of the covalent radii (Te: 1.37; S: 1.04 Å).4 As for the Rh–N bond distances associated with the Tp* ligand, trans influence exerted more strongly by the TePh ligand than the MeCN ligand results in the significantly longer Rh–N(2) and Rh–N(4) bonds trans to the TePh ligands at 2.163(6) and 2.151(6) Å, respectively, than the Rh–N(6) bond trans to the MeCN ligand at 2.064(7) Å.
| Rh–Te(1) | 2.6396(7) | Rh–Te(2) | 2.6486(7) | |
| Rh–N(2) | 2.163(6) | Rh–N(4) | 2.151(6) | |
| Rh–N(6) | 2.064(7) | Rh–N(7) | 2.005(7) | |
| N(7)–C(28) | 1.12(1) | |||
| Te(1)–Rh–Te(2) | 87.06(2) | Rh–Te(1)–C(16) | 105.1(2) | |
| Rh–Te(2)–C(22) | 106.9(2) | Rh–N(7)–C(28) | 177.4(7) | |
| N(7)–C(28)–C(29) | 177.4(9) |
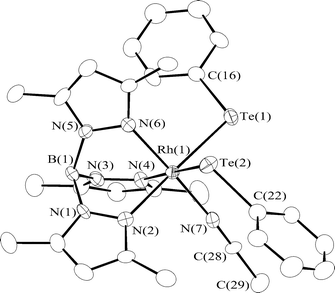 | ||
| Fig. 1 An ORTEP drawing for 2c at 30% probability level. Hydrogen atoms are omitted for clarity. | ||
For 2b, only the preliminary X-ray diffraction study was possible due to the poor quality of the crystals. Nevertheless, the similar atom-connecting scheme in the molecule has been confirmed.
Reactions of 2 with a Cp*RuII complex forming heterobimetallic complexes
We have reported previously that 2a reacts with 0.5 equiv of [(Cp*MCl)2(µ-Cl)2] (M = Rh, Cl) to give [Tp*RhCl(µ-SPh)2MCp*Cl]. Now we have found that incorporation of the Cp*RuII fragment, which is isoelectronic with the above Cp*RhIII and Cp*IrIII moieties, occurs analogously, when 2a is allowed to react with 0.25 equiv of [(Cp*Ru)4(µ-Cl)4] in THF at room temperature. The product [Tp*RhCl(µ-SPh)2RuCp*(MeCN)] (3a) in which the MeCN molecule is now bonded to the Ru site, was able to be isolated cleanly, if the product was crystallized either in a short period (< ca. 1 h) or in the presence of an excess MeCN (Scheme 1). The structure of the bimetallic core bridged by two thiolato ligands has been determined in detail by the X-ray analysis (vide infra) as shown in Fig. 2. | ||
| Fig. 2 An ORTEP drawing for 3a at 30% probability level. Hydrogen atoms are omitted for clarity. The structure and the atom numbering scheme are essentially the same for the Se analogue 3b. | ||
 | ||
| Scheme 1 | ||
The 1H NMR spectrum of 3a recorded in C6D6 shows only the unresolvable broad signals. However, when recorded in the presence of excess acetonitrile (e.g. C6D6–CD3CN in a ratio 9 : 1), the spectrum exhibits the sharp signals assignable to one Tp*, two SPh, and one Cp* groups together with the one free MeCN dissociated from the Ru site by replacement with CD3CN. With respect to the Tp* resonances, signals due to the pyrazole groups appeared as two sets with an intensity ratio of 2 : 1, while those due to the SPh groups indicated that the two Ph groups are equivalent. These spectral features in C6D6–CD3CN are diagnostic of the X-ray structure of 3a and the spectrum observed for the C6D6 solution may be explained by the presence of the equilibrium between two slowly interconverting species in C6D6, i.e.3a and the species generated by the dissociation of the coordinated MeCN from its Ru site.
Interestingly, it has turned out that when the reaction mixture of 2a with [(Cp*Ru)4(µ3-Cl)4] was kept for a longer period in the absence of MeCN, a mixture of 3a and another bimetallic complex was produced, and this new complex has been characterized by X-ray analysis (vide infra) to be the singly bridged complex [Tp*RhCl(SPh)(µ–η1-S-η6-Ph)RuCp*] (4a) depicted in Fig. 3 (Scheme 1). The 1H NMR study of 3a in C6D6 showed that in the spectra initially exhibiting only the broad peaks of 3a, the sharp signals due to 4a appeared and their intensities increased gradually. This indicates unambiguously that 3a in solution is transformed slowly into 4a even at room temperature. On the other hand, when a benzene solution of 3a was heated at reflux, conversion of 3a into 4a was completed smoothly and 4a was isolated in 41% yield in a pure form.
 | ||
| Fig. 3 An ORTEP drawing for 4a at 30% probability level. Hydrogen atoms are omitted for clarity. | ||
Reaction of the bis(selenolato) complex 2b with [(Cp*Ru)4(µ3-Cl)4] also proceeded at room temperature to afford the doubly bridged complex [Tp*RhCl(µ-SePh)2RuCp*(MeCN)] (3b). In contrast to 3a, however, the selenolato complex 3b in C6D6 shows the sharp 1H NMR resonances diagnostic of its solid state structure determined by X-ray analysis and the spectra recorded after prolonged periods were identical when kept at room temperature. These findings indicate that in 3b the MeCN ligand is bonded to Ru tightly even in solution and the core structure is stable at room temperature. On the other hand, transformation into a singly bridged complex [Tp*RhCl(SePh)(µ–η1-Se-η6-Ph)RuCp*] (4b) did also proceed for the selenolato complex 3b when more forcing conditions were employed, e.g. refluxing a benzene solution of 3b for 6 h. It is noteworthy that the tellurolato analogue [Tp*RhCl(µ-TePh)2RuCp*(MeCN)] (3c), which was obtained by similar treatment of 2c with [(Cp*Ru)4(µ3-Cl)4] and showed the 1H NMR spectral feature analogous to 3b at room temperature, did not react even at refluxing temperature. Thus, 3c dissolved in benzene was recovered quantitatively after heating at reflux for 6 h (Scheme 1).
This core rearrangement, observed for 3a even at room temperature, and for 3b under more forcing conditions, but not for 3c, may proceed because of the presence of the steric hindrance in these doubly bridged complexes. Since the hindrance between the Tp*, Cp* and Ph groups decreases in the order 3a > 3b > 3c due to the elongation of the M–E bond distances, reactivities of 3 toward conversion into less sterically hindered 4 presumably decrease in this order. More labile features of the MeCN ligand observed for 3a to generate coordinatively unsaturated Ru center also arises from the steric crowding at the Ru site that is more severe in this thiolato complex than in 3b and 3c, and is presumably ascribable to the more facile core rearrangement observed for 3a.
Closely related rearrangement of the aryloxo ligand was reported previously for the coordinatively unsaturated Cp*RuII complexes [(Cp*Ru)2(µ-OAr)2] (Ar = Ph, C6H3PriMe-2,5, C6H3But2-3,4), which are gradually converted into mononuclear complexes [Cp*Ru(η5-OAr)] at room temperature.5 By contrast, the thiolato analogue [(Cp*Ru)2(µ-SC6H3Me2-2,6)2] is stable and does not undergo the core rearrangement of this type under similar conditions.6
X-Ray structures of 3a, 3b, and 4a
The X-ray structure of 3a depicted in Fig. 2 is compares well to that of the previously described [Tp*RhCl(µ-SPh)2RhCp*(MeCN)]+.2 Thus, the RhRuS2 ring is almost planar, and the two metal centers connected by two mutually syn SPh ligands are separated by 3.6875(5) Å, which indicates the absence of any metal–metal bonding interactions. Two Ph groups of the µ-thiolato ligands are oriented toward the opposite direction to the Cp* ligand and one pyrazolyl Me group of the Tp* ligand occupies the sandwiched position between these two Ph rings.As shown in Table 2, the Rh–S bond distances at 2.354(1) and 2.355(1) Å are significantly shorter than the Ru–S bond distances at 2.432(1) and 2.437(1) Å. The Tp*Rh–S bond lengths in 3a are comparable to those in [Tp*RhCl(µ-SPh)2RhCp*(MeCN)]+ (2.357(2) and 2.371(2) Å) and other µ-thiolato and µ-hydrosulfido complexes cited in the previous paper.2 With respect to the Ru–µ-S bond lengths, those in 3a are almost identical with those in [{Cp*Ru(CO)}2(µ-SBut)2] (2.422 Å (av.))7 or somewhat longer than those in the coordinatively unsaturated Cp*RuII complexes [(Cp*Ru)2(µ-SC6H3Me2-2,6)2] (2.350 Å),6 and [{(η5-C5Me4Et)Ru}2(µ-SEt)2] (2.323 Å (av.)).8 For the related RuIII complexes, the Ru-µ-S bond lengths at 2.314 Å (av.) in [(Cp*RuCl)2(µ-SEt)2]9 and 2.319 Å (av.) in [(Cp*RuCl)2(µ-SPh)2]10 have been reported.
| 3a (E = S) | 3b (E = Se) | |
|---|---|---|
| Rh–Cl | 2.338(1) | 2.353(1) |
| Rh–E(1) | 2.354(1) | 2.4769(6) |
| Rh–E(2) | 2.355(1) | 2.4775(4) |
| Rh–N(2) | 2.128(4) | 2.154(4) |
| Rh–N(4) | 2.129(4) | 2.143(3) |
| Rh–N(6) | 2.098(3) | 2.101(3) |
| Ru–E(1) | 2.432(1) | 2.5271(5) |
| Ru–E(2) | 2.437(1) | 2.5371(7) |
| Ru–N(7) | 2.044(4) | 2.038(4) |
| Ru–C(Cp*) | 2.170(5)–2.191(5) | 2.153(7)–2.183(5) |
| N(7)–C(38) | 1.121(6) | 1.125(7) |
| Rh⋯Ru | 3.6875(5) | 3.8585(5) |
| E(1)–Rh–E(2) | 80.45(4) | 80.19(2) |
| E(1)–Ru–E(2) | 77.30(4) | 78.12(2) |
| Rh–E(1)–Ru | 100.80(4) | 100.90(2) |
| Rh–E(2)–Ru | 100.63(4) | 100.61(2) |
The Se analogue 3b has essentially the same structure as that of 3a. Pertinent bond distances and angles observed for 3b are also listed in Table 2. The Rh–Se bonds at 2.4769(6) and 2.4775(4) Å and the Ru–Se bonds at 2.5271(5) and 2.5371(7) Å are longer than the corresponding Rh–S and Ru–S bonds in 3a by ca. 0.12 and 0.10 Å, which are consistent with the difference in the covalent radii of Se at 1.17 Å and S at 1.04 Å.4 Accordingly, the Rh⋯Ru separation of 3.8585(5) Å is longer than that in 3a by ca. 0.17 Å. Well defined µ-selenolato complexes of Rh and Ru are still limited; previously reported Rh–µ-Se bond lengths are 2.481 Å (av.) in [{CpRh(SePh)2}(µ-SePh)2] and 2.502 Å (av.) in [(Cp*Rh)2(µ-SePh)3]+,11 while the Ru–µ-Se bond distances in the RuIII complexes [(Cp*Ru)2(µ-SeTol)3]Cl,12 [(Cp*RuCl)2(µ-SeFc)2] (Fc = ferrocenyl),13 [(Cp*RuCl)2(µ-SeR)2] (R = Me, Et, Prn, Pri, Ph),14 and [{CpRu(MeCN)}2(µ-SePh)2][PF6]215 are 2.456 Å (av.), 2.4218 Å, 2.415(1)–2.439(1) Å, and 2.421 Å (av.), and those in the RuI complex [{Ru(CO)2}2(µ-SePh)2(µ-Ph2PCH2PPh2)]16 are 2.524 Å (av.), respectively.
On the other hand, Fig. 3 shows the molecular structure of 4a and Table 3 summarizes the important bond lengths and angles therein. Complex 4a has a dinuclear structure containing Tp*Rh and Cp*Ru units connected by one SPh ligand, which coordinates to the Rh center at the S atom in a terminal end-on mode and to the Ru atom by the Ph ring in an η6-fashion. The Rh center has an octahedral structure with a fac-Tp*, two PhS, and one Cl ligands. The Rh–S(1) bond associated with the µ-SPh ligand is somewhat elongated from the Rh–S(2) bond in the terminal SPh ligand. On the basis of the difference in the Rh–N bond distances (Rh–N(4): 2.125(4), Rh–N(2): 2.111(4), Rh–N(6): 2.088(4) Å), the strength of the trans influence of the thiolate and Cl ligands is assumed to decrease in the order: terminal S(2)–Ph > µ-S(1)–Ph > Cl. The Rh–S–C angles are essentially the same for both the terminal and bridging SPh ligands (112.4(2) and 113.7(2)°).
| Rh–Cl | 2.359(1) | Rh–S(1) | 2.358(1) |
| Rh–S(2) | 2.331(2) | Rh–N(2) | 2.111(4) |
| Rh–N(4) | 2.125(4) | Rh–N(6) | 2.088(4) |
| Ru–C(16) | 2.276(5) | Ru–C(17) | 2.228(6) |
| Ru–C(18) | 2.200(6) | Ru–C(19) | 2.193(6) |
| Ru–C(20) | 2.170(6) | Ru–C(21) | 2.197(6) |
| Ru–C(Cp*) | 2.141(5)–2.165(6) | ||
| S(1)–Rh–S(2) | 89.98(6) | Rh–S(1)–C(16) | 113.7(2) |
| Rh–S(2)–C(22) | 112.4(2) |
Around Ru, both of the Cp* and Ph groups coordinate to the Ru atom in a π-fashion and these two planes are mutually parallel with the dihedral angle of 177.4°. The Ru–C(Cp*) bond distances differ only slightly to each other, which are in the range 2.141(5)–2.165(6) Å. By contrast, the Ru–C(Ph) bond lengths vary significantly from 2.170(6) Å of the Ru–C(20) bond up to 2.276(5) Å of the Ru–C(16) bond, where the C(20) and C(16) atoms are occupying the meta and ipso positions of the SPh ligand, respectively. Elongated Ru–C(16) distance is interpreted in terms of the contribution of the resonance structure i for 4a as shown in eqn (2).
 | (2) |
Formation of dioxygen complexes [Tp*RhCl(µ-EPh)2RuCp*(η2-O2)] (5)
By stirring the THF solutions of 3 under air at room temperature, dioxygen complexes 5 were obtained (eqn (3)). Three products 5a–5b were all isolated as single crystals in moderate yields and fully characterized by X-ray analyses. Since these three are isomorphous, an ORTEP drawing is depicted for 5a (Fig. 4) only. Important bond distances and angles in 5a–5c are listed in Table 4. | (3) |
| 5a (E = S) | 5b (E = Se) | 5c (E = Te) | |
|---|---|---|---|
| Rh–Cl | 2.3462(8) | 2.349(2) | 2.360(1) |
| Rh–E(1) | 2.3588(8) | 2.4590(7) | 2.6185(4) |
| Rh–E(2) | 2.3462(7) | 2.4645(7) | 2.6022(5) |
| Rh–N(2) | 2.100(2) | 2.115(5) | 2.154(4) |
| Rh–N(4) | 2.108(2) | 2.118(4) | 2.150(4) |
| Rh–N(6) | 2.093(2) | 2.091(5) | 2.097(3) |
| Ru–E(1) | 2.4294(7) | 2.5461(7) | 2.6900(4) |
| Ru–E(2) | 2.4316(8) | 2.5393(7) | 2.6678(5) |
| Ru–O(1) | 1.984(2) | 2.008(4) | 2.020(3) |
| Ru–O(2) | 2.004(2) | 1.994(4) | 2.007(4) |
| Ru–C(Cp*) | 2.191(3)–2.314(3) | 2.189(6)–2.292(7) | 2.205(5)–2.285(4) |
| Rh⋯Ru | 3.7073(3) | 3.8583(6) | 4.0608(5) |
| O(1)–O(2) | 1.397(3) | 1.406(6) | 1.383(6) |
| E(1)–Rh–E(2) | 79.91(3) | 80.62(2) | 80.90(1) |
| E(1)–Ru–E(2) | 76.86(2) | 77.56(2) | 78.42(1) |
| O(1)–Ru–O(2) | 41.01(9) | 41.1(2) | 40.2(2) |
| Rh–E(1)–Ru | 101.46(3) | 100.85(2) | 99.80(1) |
| Rh–E(2)–Ru | 101.77(3) | 100.89(2) | 100.80(1) |
 | ||
| Fig. 4 An ORTEP drawing for 5a at 30% probability level. Hydrogen atoms are omitted for clarity. The structures and the atom numbering schemes are essentially the same for the Se and Te analogues 5b and 5c. | ||
The structures of 5 are analogous to the corresponding 3 except for the presence of the η2-O2 ligands in place of the η1-MeCN ligands. Bonding parameters associated with the Rh(µ-EPh)2Ru core for 5a and 5b are essentially the same as those of the X-ray analyzed counterparts 3a and 3b, respectively. In 5, the Rh–E and Ru–E bond distances are elongated from 2.352 and 2.431 Å (av.) in 5a (E = S) to 2.610 and 2.679 Å (av.) in 5c (E = Te) as the covalent radii of E increase, and accordingly the Rh⋯Ru separations become greater in this order from 3.7073(3) Å in 5a to 4.0608(5) Å in 5c. To our knowledge, there are no precedented examples of the Rh–µ-TeR bond lengths, while the Ru–Te bond distances for the µ-tellurolato ligands in the RuII complexes [(Cp*Ru)2(µ-TeTol)2(µ-TolTeTeTol)]12 and [(Cp*Ru)2(µ-TeFc)2(µ-butadiene)]13 and in the RuIII complexes [(Cp*RuCl)2(µ-TeMe)2]14 were reported to be 2.676 (av.), 2.659 (av.), and 2.580(1) Å, that for the µ-TePri ligand in the alkynylvinyl cluster [Ru3Pt{µ4-C(Ph)CCCH(But)}(µ4-Te)(µ2-TePri)(CO)6(dppe)] being 2.655 Å (av.).17
The dioxygen molecule coordinates to the Ru center in an η2 manner. The O–O bond lengths in the range 1.383(6)–1.406(6) Å for these three complexes are apparently not affected by the nature of the chalcogenolato ligands and are in the range typical to the peroxo ligands (1.35–1.5 Å).18 In the IR spectra of 5, however, no bands assignable to the O2 ligands were observed, although the peroxo ligands generally show the characteristic ν(O–O) bands in the region 820–910 cm−1. As the O2 ligand is formulated as O22−, the formal oxidation state of the Ru center becomes +4 and the comparable O–O bond distances have been observed in the other RuIV peroxo complexes such as [RuH(η2-O2)(C6H4-o-py)(PPri3)2] (py = pyridyl) (1.407(3) Å),19 [Cp*Ru(η2-O2){Ni(S2N2)}]+ (S2N2 = SCH2CH2NMeCH2CH2NMeCH2CH2S) (1.371(8) Å),20 and [Cp*Ru(η2-O2)(P2)]+ (P2 = dppe: 1.398(5) Å;21P2 = dppm: 1.37(1) Å22). In contrast, the O–O distance at 1.461(5) Å in the RuII peroxo complex [Ru(η2-O2)(Ph2PNMeNMePPh2)2]23 is much longer and comparable to that in the H2O2 (1.461(3) Å).24 Reactivities of the peroxo ligands in 5 are now under investigation.
Experimental
General considerations
All manipulations were carried out under N2 using standard Schlenk techniques. Solvents were dried by common procedures and distilled under N2 before use. Complexes 125 and [(Cp*Ru)4(µ3-Cl)4]26 were prepared according to literature methods, while other reagents were commercially obtained and used as received.1H NMR spectra were recorded on a JEOL alpha-400 spectrometer (400 MHz), where the chemical shifts were referred to those of the impurities in deuterated solvents (δ 7.26 for CDCl3 and 7.15 for C6D6). IR spectra were recorded on a JASCO FT/IR-420 spectrometer and elemental analyses were performed with a Perkin-Elmer 2400 series II CHN analyzer.
![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N) cm−1.
N) cm−1.
N) cm−1. Single crystals of 3a·THF for the X-ray diffraction were grown from THF–MeCN–hexane at −20 °C.
N) cm−1.
N) cm−1.
N) cm−1.
N) cm−1.
N) cm−1. Single crystals of 3a·THF for the X-ray diffraction were grown from THF–MeCN–hexane at −20 °C.
N) cm−1.
N) cm−1.
A benzene solution (15 mL) of 3a·THF (271 mg, 0.270 mmol) was refluxed for 6 h and the resultant mixture was filtered off and the orange solid remaining was extracted with CH2Cl2. Addition of hexane to the concentrated extract afforded 4a as orange–red crystals (133 mg, 55% yield).
Crystallography
Single crystals of 2b, 3a·THF, 3b·2.625THF, 4a·THF, 5a·THF, 5b·THF, and 5c·THF were sealed in glass capillaries under argon and mounted on a Rigaku Mercury-CCD diffractometer equipped with a graphite-monochromatized Mo-Kα source. All diffraction studies were performed at 23 °C, whose details are listed in Table 5.| 2c | 3a·THF | 3b·2.625THF | 4a·THF | 5a·THF | 5b·THF | 5c·THF | |
|---|---|---|---|---|---|---|---|
| a R1 = Σ‖ Fo| − |Fc‖/Σ|Fo|. Based on the data with I > 2σ(I). b wR2 = [Σw(Fo2 − Fc2)2/Σ w(Fo2)2]1/2. Based on all data. c GOF = [Σ w(|Fo| − |Fc|)2/{(no. observed) − (no. variables)}]1/2. | |||||||
| Formula | C29H35N7BTe2Rh | C43H58N7OBS2ClRhRu | C49.5H71N7O2.625BSe2ClRhRu | C41H55N6OBS2ClRhRu | C41H55N6O3BS2ClRhRu | C41H55N6O3BSe2ClRhRu | C41H55N6O3BTe2ClRhRu |
| Formula weight | 850.56 | 1003.34 | 1214.39 | 962.28 | 994.28 | 1088.08 | 1185.36 |
| Space group | P21/a (no. 14) | P21/c (no. 14) | P-1 (no. 2) | P21/c (no. 14) | P21/c (no. 14) | P21/c (no. 14) | P21/c (no. 14) |
| a/Å | 8.189(3) | 11.055(2) | 12.159(2) | 12.797(5) | 11.224(2) | 11.233(2) | 11.439(2) |
| b/Å | 23.512(9) | 19.403(3) | 13.757(2) | 17.828(6) | 19.355(4) | 19.361(3) | 12.020(2) |
| c/Å | 17.625(7) | 22.302(3) | 18.045(2) | 20.634(8) | 20.168(4) | 20.392(3) | 32.913(7) |
| α/° | 90 | 90 | 72.811(5) | 90 | 90 | 90 | 90 |
| β/° | 97.054(2) | 90.538(7) | 78.756(6) | 106.032(2) | 92.093(1) | 92.5895(7) | 96.2910(8) |
| γ/° | 90 | 90 | 78.868(6) | 90 | 90 | 90 | 90 |
| U/Å3 | 3368(2) | 4783(1) | 2798.4(6) | 4524(3) | 4379(2) | 4431(1) | 4498(2) |
| Z | 4 | 4 | 2 | 4 | 4 | 4 | 4 |
| µ/cm−1 | 22.33 | 8.42 | 19.56 | 8.86 | 9.22 | 24.60 | 20.76 |
| Total reflections | 7981 | 11380 | 13051 | 10747 | 10309 | 10542 | 10059 |
| Data with I > 2σ(I) | 3551 | 6275 | 8494 | 4580 | 7867 | 5824 | 7261 |
| Variables | 399 | 575 | 656 | 528 | 563 | 563 | 563 |
| R1a | 0.050 | 0.042 | 0.043 | 0.048 | 0.037 | 0.045 | 0.037 |
| wR2b | 0.163 | 0.134 | 0.134 | 0.156 | 0.112 | 0.139 | 0.114 |
| GOFc | 1.02 | 1.00 | 1.06 | 1.02 | 1.02 | 1.03 | 1.00 |
Structure solution and refinements were carried out by using the CrystalStructure program package.27 The positions of the non-hydrogen atoms were determined by Patterson methods (PATTY)28 and subsequent Fourier synthesis (DIRDIF 99).29 These were refined with anisotropic thermal parameters by full-matrix least-squares techniques except for compounds 3b and 4a in which 5 and 1 atoms, respectively, were refined isotropically. Hydrogen atoms bound to the B atoms were found in the Fourier maps and refined isotropically, while all other hydrogens were placed at the calculated positions and included at the final stages of the refinements with fixed parameters. For 4a·THF, solvating THF molecule is occupying the disordered positions, which has been modeled as two differently oriented five-membered rings overlapping each other with the occupancies 1 : 1.
For 2b, only the preliminary results were obtained. Crystallographic data are as follows. 2b: C29H35N7BSe2Rh, M = 753.28, space group P21/n (no. 14), a = 19.762(8), b = 8.354(3), c = 19.128(8) Å, β = 101.213(6)°, U = 3098(2) Å3, Z = 4, µ = 29.32 cm−1, 5848 data (I > 3σ(I)), 396 variables, R1 = 0.083, wR2 = 0.170. Attempts to lower the R values to the satisfactory levels were unsuccessful.
CCDC reference numbers 265326–265332.
See http://dx.doi.org/10.1039/b503153m for crystallographic data in CIF or other electronic format.
Acknowledgements
This work was supported by Grant-in-Aid for Scientific Research on Priority Areas (No. 14078206, “Reaction Control of Dynamic Complexes”) from the Ministry of Education, Culture, Sports, Science and Technology, Japan and by CREST of JST (Japan Science and Technology Agency).References
- (a) S. Trofimenko, Scorpionates, Imperial College Press, London, 1999 Search PubMed; (b) C. Slugovc, R. Schmid and K. Kirchner, K., Coord. Chem. Rev., 1999, 185–186, 109 CrossRef CAS; (c) N. Kitajima and W. B. Tolman, Prog. Inorg. Chem., 1995, 43, 419 CAS; (d) S. Trofimenko, Chem. Rev., 1993, 93, 943 CrossRef CAS.
- H. Seino, T. Yoshikawa, M. Hidai and Y. Mizobe, Dalton Trans., 2004, 3593 RSC.
- J. Arnold, Prog. Inorg. Chem, 1995, 43, 353 CAS.
- Lange's Handbook of Chemistry, ed. by J. A. Dean, McGraw-Hill, Inc., New York, 1985, 13th edn, Table 3–10 Search PubMed.
- K. Bücken, U. Kölle, R. Pasch and B. Ganter, Organometallics, 1996, 15, 3095 CrossRef.
- A. Takahashi, Y. Mizobe, H. Matsuzaka, S. Dev and M. Hidai, J. Organomet. Chem., 1993, 456, 243 CrossRef CAS.
- A. Hörnig, C. Rietmann, U. Englert, T. Wagner and U. Kölle, Chem. Ber., 1993, 126, 2609 CrossRef.
- U. Kölle, C. Rietmann and U. Englert, J. Organomet. Chem., 1992, 423, C20 CrossRef.
- K. Hashizume, Y. Mizobe and M. Hidai, Organometallics, 1996, 15, 3303 CrossRef CAS.
- T. Kondo, S. Uenoyama, K. Fujitaand and T. Mitsudo, J. Am. Chem. Soc., 1999, 121, 482 CrossRef CAS.
- M. Herberhold, M. Keller, W. Kremnitz, T. Daniel, W. Milius, B. Wrackmeyer and H. Nöth, Z. Anorg. Allg. Chem., 1998, 624, 1324 CrossRef CAS.
- H. Matsuzaka, T. Ogino, M. Nishio, M. Hidai, Y. Nishibayashi and S. Uemura, J. Chem. Soc., Chem. Commun., 1994, 223 RSC.
- H. Matsuzaka, J.-P. Qü, T. Ogino, M. Nishio, Y. Nishibayashi, Y. Ishii, S. Uemura and M. Hidai, J. Chem. Soc., Dalton Trans., 1996, 4307 RSC.
- (a) Y. Nishibayashi, H. Imajima, G. Onodera, Y. Inada, M. Hidai and S. Uemura, Organometallics, 2004, 23, 5100 CAS; (b) Y. Nishibayashi, H. Imajima, G. Onodera, M. Hidai and S. Uemura, Organometallics, 2004, 23, 26 CrossRef CAS.
- E. Becker, K. Mereiter, R. Schmid and K. Kirchner, Organometallics, 2004, 23, 2876 CrossRef CAS.
- S. E. Kabir, S. J. Ahmed, Md. I. Hyder, Md. A. Miah, D. W. Bennett, D. T. Haworth, T. A. Siddiquee and E. Rosenberg, J. Organomet. Chem., 2004, 689, 3412 CrossRef CAS.
- L. J. Farrugia, N. MacDonald and R. D. Peacock, Polyhedron, 1998, 17, 2877 CrossRef CAS.
- L. Vaska, Acc. Chem. Res., 1976, 9, 175 CrossRef CAS.
- J. Mattes, S. Gründemann, A. Toner, Y. Guari, B. Donnadieu, J. Spandl, S. Sabo-Etienne, E. Clot, H.-H. Limbach and B. Chaudret, Organometallics, 2004, 23, 1424 CrossRef.
- M. A. Reynolds, T. B. Rauchfuss and S. R. Wilson, Organometallics, 2003, 22, 1619 CrossRef CAS.
- K. Kirchner, K. Mauthner, K. Mereiter and R. Schmid, J. Chem. Soc., Chem. Commun., 1993, 892 RSC.
- G. Jia, W. S. Ng, H. S. Chu, W.-T. Wong, N.-T. Yu and I. D. Williams, Organometallics, 1999, 18, 3597 CrossRef CAS.
- J. Shen, E. D. Stevens and S. P. Nolan, Organometallics, 1998, 17, 3875 CrossRef CAS.
- J.-M. Savariault and M. S. Lehmann, J. Am. Chem. Soc., 1980, 102, 1298 CrossRef CAS.
- K. Ohta, M. Hashimoto, Y. Takahashi, S. Hikichi, M. Akita and Y. Moro-oka, Organometallics, 1999, 18, 3234 CrossRef CAS.
- C. White, A. Yate and P. M. Maitlis, Inorg. Synth., 1992, 29, 228 CAS.
- Crystal Structure 3.00: Crystal Structure Analysis Package, Rigaku and Rigaku/MSC, 2000–2002 Search PubMed; CRYSTALS Issue 10: D. J. Watkin, C. K. Prout, J. R. Carruthers and P. W. Betteridge, Chemical Crystallography Laboratory: Oxford, UK Search PubMed.
- PATTY: P. T. Beurskens, G. Admiraal, G. Beurskens, W. P. Bosman, S. Garcia-Granda, R. O. Gould, J. M. M. Smits and C. Smykall, The DIRDIF program system; Technical Report of the Crystallography Laboratory: University of Nijmegen, Nijmegen, The Netherlands, 1992 Search PubMed.
- DIRDIF99: P. T. Beurskens, G. Admiraal, G. Beurskens, W. P. Bosman, R. de Gelder, R. Israel and J. M. M. Smits, The DIRDIF-99 program system; Technical Report of the Crystallography Laboratory: University of Nijmegen, Nijmegen, The Netherlands, 1999 Search PubMed.
| This journal is © The Royal Society of Chemistry 2005 |