Formation of a cytochrome c–nitrous oxide reductase complex is obligatory for N2O reduction by Paracoccus pantotrophus†
Tim
Rasmussen
ab,
Thomas
Brittain
c,
Ben C.
Berks
d,
Nicholas J.
Watmough
a and
Andrew J.
Thomson
*b
aCentre for Metalloprotein Spectroscopy and Biology, School of Biological Sciences, University of East Anglia, Norwich, UK NR4 7TJ
bCentre for Metalloprotein Spectroscopy and Biology, School of Chemical Sciences and Pharmacy, University of East Anglia, Norwich, UK NR4 7TJ. E-mail: a.thomson@uea.ac.uk; Fax: +44 1603 593051; Tel: +44 1603 593051
cSchool of Biological Sciences, University of Auckland, Auckland, New Zealand
dDepartment of Biochemistry, University of Oxford, South Parks Road, Oxford, UK OX1 3QU
First published on 23rd September 2005
Abstract
Nitrous oxide reductase (N2OR) catalyses the final step of bacterial denitrification, the two-electron reduction of nitrous oxide (N2O) to dinitrogen (N2). N2OR contains two metal centers; a binuclear copper center, CuA, that serves to receive electrons from soluble donors, and a tetranuclear copper-sulfide center, CuZ, at the active site. Stopped flow experiments at low ionic strengths reveal rapid electron transfer (kobs = 150 s−1) between reduced horse heart (HH) cytochrome c and the CuA center in fully oxidized N2OR. When fully reduced N2OR was mixed with oxidized cytochrome c, a similar rate of electron transfer was recorded for the reverse reaction, followed by a much slower internal electron transfer from CuZ to CuA (kobs = 0.1–0.4 s−1). The internal electron transfer process is likely to represent the rate-determining step in the catalytic cycle. Remarkably, in the absence of cytochrome c, fully reduced N2OR is inert towards its substrate, even though sufficient electrons are stored to initiate a single turnover. However, in the presence of reduced cytochrome c and N2O, a single turnover occurs after a lag-phase. We propose that a conformational change in N2OR is induced by its specific interaction with cytochrome c that in turn either permits electron transfer between CuA and CuZ or controls the rate of N2O decomposition at the active site.
Introduction
Nitrous oxide (N2O) along with carbon dioxide (CO2) and methane is one of the three most important greenhouse gases. The largest source of man-made N2O emissions is agriculturally derived because soluble nitrate (NO3−) and nitrite (NO2−) ions, in the form of artificial fertilizers, are converted by some bacteria and fungi to atmospheric nitrogen in a process known as denitrification, in the course of which N2O gas, an intermediate, escapes from cells.1,2 Denitrification requires four specialist enzyme activities to catalyze the sequential conversion of NO3− → NO2− → NO → N2O → N2.3,4 Of these the final step of denitrification, the 2-electron reduction of nitrous oxide to dinitrogen is catalysed by nitrous oxide reductase (N2OR) an enzyme found in bacteria but not apparently in denitrifying fungi.5 A detailed investigation of the mechanism of N2OR would help our understanding of this activity and how it might be harnessed in remedial processes aimed at limiting N2O emissions.N2OR is a homodimer that is located in the bacterial periplasm. Recent X-ray structures of the enzymes from Pseudomonas nautica and Paracoccus denitrificans have revealed that each subunit (MW = 65 kDa) is organized into two domains6,7 (Figs. 1a and 1b). One cupredoxin-like domain contains a binuclear copper center known as CuA. The CuA center can undergo a one-electron redox change and hence has a function similar to that in the well-known aa3-type cytochrome c oxidases (CcO) where it serves to receive an electron from soluble cytochromes c8 (Fig. 1c). For example, in Rhodobacter capsulatus, cytochrome c2 is essential for electron transfer to N2OR.9 Moreover, studies on Rhodobacter sphaeroides,10Wolinella succinogenes11 and Paracoccus pantotrophus12–14 have established the cognate periplasmic cytochromes c as natural electron donors to N2OR. At least in vitro, the cupredoxin pseudoazurin can also function as an alternative electron donor to N2OR isolated from P. pantotrophus.14 In the closely related organism P. denitrificans deletion of the cytochrome c550 gene does not compromise its ability to reduce nitrous oxide suggesting that pseudoazurin may also be a natural electron donor to N2OR in vivo.15
 | ||
| Fig. 1 Structure of nitrous oxide reductase and its copper clusters. (a) Structure of one subunit of the homodimeric N2OR. (b) The subunit structure of N2OR showing the head to tail arrangement that brings the electron storage centre CuA to within ca. 10 Å of the catalytic cluster CuZ in the other subunit. (c) The molecular structures and redox states of the CuA and CuZ clusters. | ||
The second domain comprises a 7-bladed propeller of β-sheets that contains the catalytic site,7 a unique [Cu4S] center, called CuZ, Figs. 1a and 1c. The original structural report at 2.9 Å resolution of the enzyme from P. nautica, failed to assign the μ4 ligand in CuZ to inorganic sulfide. A combination of elemental analysis and resonance Raman spectroscopy of a 34S enriched sample showed clearly that sulfur was a ligand to CuZ.16 Recently a higher resolution (1.6 Å) structure of the enzyme from P. denitrificans in which the electron density at the CuZ center is better resolved confirms this assignment.6 The distance between the CuA and CuZ centers within a single subunit is greater than 30 Å, a distance that precludes physiologically relevant rates of intra-subunit electron transfer. However, the two subunits are orientated “head to tail” such that the CuA center in one subunit lies only 10 Å from the CuZ center in the second ensuring that pairs of redox centers in opposite subunits form the catalytically competent unit.
A pre-requisite to understanding the catalytic cycle of N2OR is the characterization of its redox properties. It is well established that CuA is capable of carrying only one electron. However, of the five oxidation levels that can be written for CuZ, assuming each copper ion can adopt either +I or +II states, only a diamagnetic state [CuII2CuI2S]4+ and a S = ½ paramagnetic form [CuII1CuI3S]3+ have been well characterized17,18 (Fig. 1c). Recently reported work19 established that the enzyme will accept two electrons per subunit when purified under anaerobic conditions but only one when prepared aerobically. The difference between the two forms of the enzyme depends on the state of the CuZ center.19 An optically monitored equilibrium redox titration of the anaerobically prepared form of the enzyme showed that, in the absence of substrate, two electrons are transferred, each in a Nernstian process at +260 mV and +60 mV, the former corresponding to the addition of one electron to CuA and, the latter, of one electron to CuZ.19 The enzyme can be prepared with each monomer in a fully oxidized, one-electron reduced (in which only CuA is reduced), or two-electron reduced state (in which both CuA and CuZ have accepted one electron). The one-electron reduced form is prepared with ascorbate, whereas the two-electron reduced state requires exposure to excess dithionite. Under these conditions, one-electron reduction of CuZ to the [Cu3ICu1II]3+ state takes about one hour.20 The steady state activities towards N2O reduction of all these forms of the enzyme, when assayed with HH cytochrome c are similar at 0.034 U mg−1.14 However, use of the artificial electron donors, benzyl viologen and methyl viologen rather than the natural electron donors, in steady-state kinetic measurements results in increased activity of the enzyme to ca. 5 U mg−1 which increases further over the course of several hours. For the P. denitrificans enzyme we observe a rise from ca. 5 to ca. 50 U mg−1 over 100 minutes. The activity of the Achromobacter cycloclastes N2OR reached 125 U mg−1 after 8 hours21 and in the case of P. nautica enzyme a maximum specific activity of 275 U mg−1 was attained after 60 min.22 During this activation process the EPR signal characteristic of CuZ is lost suggesting that the cluster can be reduced by two electrons to the all Cu(I) state, [CuI4S]2+.21,22 Hence, the enzyme can be artificially super-reduced to a state carrying three electrons per subunit, one on CuA and two on CuZ. However, this activation process is slow and requires a much more powerful reductant than is available in the periplasm of a Gram-negative bacterium.
Mechanistic studies are required to establish the relevance of this super-reduced state of CuZ to enzymatic catalysis and to determine whether it is formed in small amounts under turnover conditions. Few mechanistic studies have been reported to date and these have used the artificial electron donors, benzyl viologen and methyl viologen in steady-state kinetic measurements,23–27 rather than the natural electron donors that are needed if conclusions are to be drawn about the catalytic cycle. To date the most detailed kinetic study using cytochrome c as the electron donor, has been performed with N2OR from W. succinogenes.11 However, the kinetic behaviour of N2OR from this organism may not be typical since N2OR from W. succinogenes has an additional domain that contains a c-type heme not found in N2OR from any other source.26,28
This paper presents the first kinetic experiments on N2OR carried out under pre-steady-state conditions. We have investigated the reactions of N2OR in the oxidized, one-electron and two-electron reduced states with HH cytochrome c and N2O, as well as carrying out single turnover experiments on the enzyme. HH cytochrome c is an efficient electron donor to several other enzymes of denitrification that have been purified from P. pantotrophus, for example, cytochrome cd1 nitrite reductase.29 Moreover, HH cytochrome c is reported to give a higher steady-state rate of N2O reduction with N2OR from P. pantotrophus compared with cytochromes c from other sources.14 The results of these experiments allow us to propose a model of the catalytic cycle for N2OR.
Experimental
The preparation of N2OR from P. pantotrophus has been described elsewhere.19 For the results reported here, the anaerobic form of the enzyme was used.19 The experiments described here were carried out with the enzyme in a single redox state generated by incubating N2OR with 2 mM ferricyanide, ascorbate or dithionite for two hours to form the oxidized, one-electron and two-electron reduced forms, respectively. Excess oxidant or reductant were removed by buffer exchange using Microcon-30 centrifuge filter devices (Millipore). HH cytochrome c, purchased from Sigma and further purified as described elsewhere30 was oxidized with 2 mM ferricyanide, or reduced with 2 mM ascorbate, and removed from excess reactants with Microcon-10 centrifuge filter devices. All operations were performed in a glove box under anaerobic conditions. The methyl viologen-linked N2O reductase activity was measured spectrophotometrically as described elsewhere.24UV/Vis stopped flow experiments were performed with an Applied Photophysics Bio-Sequential DX.17 MV spectrophotometer or a High Tech SF 61DX2 instrument in a glove box, both with a 1 cm path length cell. Absorption changes were detected either with a photomultiplier at a single wavelength or a photodiode array from 430 to 1000 nm. In the latter case, a cut off filter at 420 nm was employed in order to avoid photo-oxidation of HH cytochrome c. All solutions for the stopped flow experiments were buffered with 2.5 mM Tris/HCl, pH 7.5 and were made anaerobic by flushing with nitrogen gas. In the single-turnover experiments, either the N2OR solution or the HH cytochrome c solution was saturated with N2O. Further details are described in the figure legends. The kinetic data were analyzed with the programs Origin (Microcal) and TableCurve 2D (Jandel). Rate constants were determined by fitting the experimental time courses to exponential or second order functions as appropriate. Non-linear concentration dependence data were fitted to a hyperbolic function by non-linear least squares procedures. Computer simulations of single turnover time courses were performed using the simulation program Gepasi.31
Results
The rate of electron transfer between reduced HH cytochrome c and oxidized N2OR was determined in the absence of N2O. Cytochrome c oxidation was monitored at 449 nm (Δε = 10.7 mM−1 cm−1) and redox changes in CuA were monitored at 830 nm (Δε = 2.0 mM−1 cm−1), a region of the spectrum in which there is no contribution from cytochrome c (Fig. 2). In these experiments no reduction of CuZ was observed, presumably because the reduction potential of CuZ, at +60 mV, is approximately 200 mV lower than the reduction potentials of either CuA or HH cytochrome c.19 Approximately 20% of the optical density change could not be resolved in our experiments since it occurs within the dead time of the instrument (ca. 2 ms). The magnitude of this absorbance change was determined from control experiments in which either cytochrome c or N2OR solutions were mixed with anaerobic buffer. The extent of oxidation of cytochrome c was calculated from the absorbance change at 449 nm, corrected for the contributions from CuA and from the reaction occurring within the dead time. The amount of cytochrome c that is oxidized is dependent upon the concentration of cytochrome c. The concentration dependence is described by a hyperbolic curve (Fig. 3), consistent with an equilibrium being established between cytochrome c and CuA. The apparent reduction potential of the solution at equilibrium was determined from the ratio of the concentrations of oxidized to reduced cytochrome c using eqn. (1):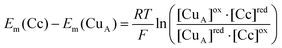 | (1) |
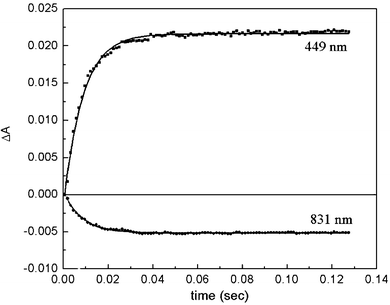 | ||
| Fig. 2 Electron-equilibration between HH cytochrome c and CuA. In a stopped flow experiment, 3.9 µM oxidized N2OR monomer was mixed with a 9-fold excess of reduced HH cytochrome c. Time courses at 449 nm and 831 nm, are shown, which monitor the redox state of HH cytochrome c, and CuA, respectively. Additionally, single-exponential fits (kobs = 150 s−1) to the data are shown. | ||
 | ||
| Fig. 3 Dependence of the electron-equilibration between HH cytochrome c and CuAon the relative HH cytochrome c concentration. The number of electrons donated by HH cytochrome c was calculated from the absorbance change at 449 nm (squares) and 551 nm (triangles), and the electrons accepted by CuA from the change at 831 nm (circles). These data are fit to eqn. (1) (solid line). | ||
 | ||
| Fig. 4 Dependence of the rate of electron-equilibration between HH cytochrome c and CuAon the ionic strength. 4.5 µM oxidized N2OR was mixed in a stopped flow experiment with 15 µM reduced HH cytochrome c at different NaCl concentrations in 2.5 mM TrisCl pH 7.5 buffer. Absorbance changes were monitored at 449 nm (squares) and at 831 nm (circles). | ||
In other experiments, oxidized HH cytochrome c was rapidly mixed with two-electron reduced N2OR in which both CuA and CuZ are each reduced by a single electron. Under these circumstances we observed a rapid electron transfer between CuA and cytochrome c (Fig. 5a) with a rate (150 s−1) very similar to that seen in the cytochrome c oxidation experiment. However, CuZ, detected at 691 nm, was oxidized with a 1000-fold slower rate. The consequent slow intra-molecular electron transfer from CuZ to CuA leads to the slow, rate limited second phase of cytochrome c reduction (Fig. 5b). The rate of the latter phase does not vary with the concentration of HH cytochrome c but does vary slightly from preparation to preparation of N2OR (kobs = 0.1–0.4 s−1).
 | ||
| Fig. 5 Reaction of oxidized HH cytochrome c with reduced N2OR. 13 µM of dithionite-reduced N2OR was mixed with 21 µM oxidized HH cytochrome c. The absorption changes recorded over a short (a) and a long (b) time scale are shown. Absorption changes were recorded at representative wavelengths: 449 nm (HH cytochrome c), 691 nm (CuZ), and 831 nm (CuA) are shown. | ||
If either oxidized or one-electron-reduced N2OR is incubated with N2O, no changes in the oxidation state of the metal centers can be detected by UV/Vis spectroscopy. Two-electron reduced N2OR shows only a very slow oxidation of CuZ by N2O in spite of possessing two electrons which are sufficient for the reaction to proceed. However, when an excess of one-electron reduced N2OR was mixed with reduced HH cytochrome c in the presence of high N2O concentrations a single turnover occurred which was detected by the simultaneous oxidation of HH cytochrome c and CuA (Fig. 6). The time course for the reaction shows a lag-phase at all wavelengths studied, as previously reported in steady-state experiments.14 With increasing HH cytochrome c concentrations, the turnover is saturated when [HH cytochrome c] > 12 µM with kobs ≤ 1.5 × 10−2 s−1 for the slower step. The single turnover rate was found to be independent of the N2O concentration in the range of 0.5 to 12 mM in these experiments.
 | ||
| Fig. 6 Single turnover of semi-reduced N2OR with N2O and HH cytochrome c. The reaction of 9 µM semi-reduced N2OR when mixed in a stopped flow apparatus with 7 µM reduced HH cytochrome c and 12 mM N2O was monitored at 449 nm (circles) and 831 nm (triangles). Simulations of this reaction based on Scheme 1 are shown (solid lines). This started with the semi-reduced form. Steps 1 to 2, 7 to 8 and 8 to 1 were not included. The rates of other steps were identified either from other experiments or determined by best fitting of the data. The outcome was k2 = 150 s−1, k−2 = 150 s−1, k3 = 3 × 10−3 s−1, k−3 = 0.15 s−1, k4 = 1 × 106 s−1 M−1, k−4 = 5 s−1, k5 = 1 × 105 s−1, k−5 = 200 s−1, k6 = k2, k−6 = k−2. k7 and k8 must be fast since they do not affect the simulation. A rate of 1 × 105 s−1 was used. | ||
When the oxidized form of N2OR was used, rather than the one-electron reduced form, fast electron equilibration between reduced HH cytochrome c and CuA, as described above, was also observed. The rate of turnover, however, was about the same as that seen in experiments using two-electron reduced N2OR.
A single turnover experiment was also performed in three other ways. HH cytochrome c was pre-mixed with N2O and rapidly mixed with N2OR, was pre-mixed with N2OR and then rapidly mixed with N2O, and finally N2OR was pre-mixed with N2O and rapidly mixed with HH cytochrome c. No differences were observed between the kinetic behaviour under any of these conditions.
Discussion
In this study we have measured the electron transfer rates between cytochrome c, an electron donor that is a homologue of the physiological electron donor cytochrome c550, and the CuA group of the enzyme, N2OR. We have also studied the ionic dependence of the interaction between these proteins. Initial studies of the electron transfer process from reduced cytochrome c to N2OR show that it is capable of reducing only the CuA center, in a process that is rate-limited at low ionic strengths by inter-molecular electron-transfer between the proteins (kobs = 150 s−1) rather than rate of association of the two proteins.Interestingly, the rate of inter-molecular electron transfer that we observe is low in comparison with the similar reaction between cytochrome c and CcO.33,34 It has been reported that in the case of CcO a conserved tryptophan residue, localized on the CcO surface close to the CuA, is crucial for a fast electron transfer reaction.34 Mutation of this residue substantially lowers the electron transfer rate to values comparable to the cytochrome c–N2OR reaction. Structural differences between CcO and N2OR could therefore account for the lower transfer rates to N2OR. Electron transfer between cytochrome c and N2OR is, however, still much faster than the observed rate of turnover (described later) and, therefore, not rate limiting. Experimental investigations of electron transfer from reduced N2OR to oxidized cytochrome c indicate the presence of two kinetic processes. The first of these processes, that is the electron transfer from CuA to cytochrome c, occurs at a concentration independent rate of 150 s−1. The close correspondence of this rate with that of the opposed reduction rate is consistent with the essentially identical reduction potentials of the CuA and cytochrome c redox centers reported above. The second, slower process involves the intra-molecular electron transfer from the reduced CuZ center to the newly oxidized CuA center. This reaction occurs with a first order rate constant of 0.1 s−1. Using this rate constant and the equilibrium constant derived from the measured difference in reduction potential between the CuZ and CuA centers we initially determined the intra-molecular rate of electron transfer from CuA to CuZ to be approximately 1 × 10−4 s−1. Since CuA is oxidized faster than CuZ, the rate of intra-molecular electron transfer between these groups must be rate limiting, rather than the inter-molecular electron transfer to cytochrome c. Moreover, as further reduction of cytochrome c in the second phase occurs at the same rate as the oxidation of CuZ, neither the dissociation of the cytochrome c reduced first during the fast phase, nor the association of a second oxidized cytochrome c, which can accept a second electron, can be rate limiting.
These reactions were all studied in the absence of substrate, N2O. Next we followed the reaction of the enzyme with N2O in three different oxidation levels, oxidized, one-electron and two-electron reduced. Simple addition experiments indicate that none of the forms, oxidized, one-electron or two-electron reduced, are capable of reducing the substrate, N2O at a significant rate. Hence, neither states of CuZ, [CuII2CuI2S]4+ or [CuII1CuI3S]3+, can react with N2O even in the presence of excess chemical reductant. Moreover, ligand exchange studies indicate that N2O does not bind tightly with one-electron reduced N2OR.
In contrast a single turnover of the one-electron reduced form of N2OR with substrate N2O is observed if cytochrome c is used as the electron donor. The kinetics of oxidation of both cytochrome c and CuA are similar and both show a lag-phase (Fig. 6). No absorption change in the range from 430 to 1000 nm could be associated with any intermediate species during this lag-phase nor could we detect reduction of CuZ during this period. This might simply be due to very low concentrations of the intermediate species. The insensitivity displayed by the reaction kinetics to the order of mixing suggests that the association of the substrate is not rate limiting, a fact confirmed by the lack of dependence of the rate on the concentration of N2O.
We have combined all of these findings to construct a possible scheme for the action of N2OR with its soluble redox partner proteins (Scheme 1). There are two important features of this scheme. First, we envisage N2O binding to the CuZ center when it is in the [CuII1CuI3S]3+ state within the one-electron reduced (CuA oxidized-CuZ reduced) form of the enzyme that forms at low concentrations during turnover (intermediate 4 of Scheme 1). Secondly, we propose that cytochrome c must form a complex with N2OR to allow transfer of the second electron from CuA to CuZ to form the super-reduced [CuI4S]2+ state that can reduce the substrate. Note that the one-electron reduced form would not be populated in the presence of dithionite. As a consequence we propose that the two-electron reduced enzyme formed in the reaction with dithionite is probably of no biological significance. Initial computer simulations employing Scheme 1 and the intra-molecular electron transfer rate from CuA to CuZ, determined from the independently measured redox potential for CuZ, yielded time courses for turnover which closely matched the form of the experimental data. However, the amplitude of the theoretical time courses only matched those of the experimental data if the intra-molecular electron transfer rate was 3 × 10−3 s−1. In simple terms, the rate calculated from the redox potential data is too small to account for the rate of N2O reduction determined experimentally. In order to account for the observed rate of N2O reduction the redox potential of the CuZ centre in the enzyme during turnover must be 150 mV rather than 60 mV. This discrepancy could arise in two ways. Firstly, the reported redox potential for the CuZ centre was determined from the oxidation of the two-electron reduced enzyme in the absence of cytochrome c and so the value measured may not be appropriate for the enzyme during turnover. Alternatively as we have described above, evidence exists that the enzymic activity of N2OR is affected by complex formation with cytochrome c. Whether this gating function derives directly from some conformationally coupled change in the structural environment of CuZ simultaneous with complex formation, or whether the redox potential change is consequent on the formation of a semi-reduced conformation cannot be determined at this time.
 | ||
| Scheme 1 Proposed catalytic cycle of N2OR in the presence of reductant, cytochrome c, and substrate N2O. The rate for the forward reaction of the internal ET (k2) was calculated from k−2 and Keq, based on the experimental midpoint potentials of CuA and CuZ in the absence of HH cytochrome c. It is assumed that the second one-electron reduction of CuA has a similar rate to the first. | ||
Additionally, the question remains as to how N2O is activated by the enzyme in the two-electron reduced state when the two electrons required to reduce the substrate are shared between two redox sites, CuARed and CuZRed, about 10 Å apart.6,7 One possibility is that a one-electron intermediate [N2O]−1 is formed allowing a second electron to be transferred from CuARed to an intermediate CuZ–[N2O]−1. The second possibility is that reduced CuZ can be further reduced in the presence of both substrate (N2O) and cytochrome c to a super-reduced, all cuprous state, [CuI4S]2+. This state would be expected to be diamagnetic (CuI = d10, S = 0) and lacking the intense charge-transfer bands of the partially oxidized redox-states of CuZ.21 We favor a role for this species in our reaction scheme even though it would be formed transiently at very low concentrations and would be difficult to detect spectroscopically.
The participation of super-reduced CuZ would be consistent with recent reports that indicate that this species is found in the enzyme after prolonged reduction with either methyl-or benzyl-viologen and that it will directly reduce N2O.21,22 In steady-state assays using these artificial electron donors the rate of turnover is significantly enhanced if the reaction is initiated with fully reduced N2OR,21 although, in the oxidizing environment of the bacterial periplasm there are not any known redox proteins with a sufficiently low reduction potential (> −400 mV) that can serve as electron donors to reduce fully the CuZ center.
It has previously been shown that steady-state rates observed with cytochrome c, or pseudoazurin, as electron donor (ca. 0.04 s−1) are much lower than in experiments with viologens (1–10 s−1) and depend on the electron donating protein.11,14 It was, therefore, concluded that electron donation is probably the rate-limiting step in the catalytic cycle of N2OR. Direct analysis of this reaction does not support this conclusion. In fact, the electron donation, including association and dissociation of cytochrome c, is fast compared to the turnover. However, we have found a slow internal electron transfer of about 0.1 s−1 in the exergonic direction from CuZ to CuA. In order to explain the fast turnover with viologens one must assume that these electron donors bypass the electron-transfer path used with cytochrome c and that the decomposition of the substrate N2O is not rate limiting just as in the case of the cytochrome c–N2O turnover.
This argues that a conformational change in N2OR is induced by its specific interaction with cytochrome c which in turn permits electron transfer between CuA and CuZ to allow formation of the super-reduced [CuI4S]2+ form of CuZ. The postulated complex has not yet been confirmed by independent experiments. This model will form the basis for further experimentation aimed at understanding why intra-molecular electron transfer in N2OR appears to be so highly controlled in turnover.
Acknowledgements
We thank the UK Research Councils EPSRC and BBSRC for support of this work through grants 83/B08413 and 83/B11958.References
- G. P. Robertson, E. A. Paul and R. R. Harwood, Science, 2000, 289, 1922–1925 CrossRef CAS.
- N. Takaya, M. A. Catalan-Sakairi, Y. Sakaguchi, I. Kato, Z. Zhou and H. Shoun, Appl. Environ. Microbiol., 2003, 69, 3152–3157 CrossRef CAS.
- B. C. Berks, S. J. Ferguson, J. W. B. Moir and D. J. Richardson, Biochim. Biophys. Acta, 1995, 1232, 97–173 CrossRef CAS.
- D. J. Richardson and N. J. Watmough, Curr. Opin. Chem. Biol., 1999, 3, 207–219 CrossRef CAS.
- H. Shoun and T. Tanimoto, J. Biol. Chem., 1991, 266, 11078–11082 CAS.
- T. Haltia, K. Brown, M. Tegoni, C. Cambillau, M. Saraste, K. Mattila and K. Djinovic-Carugo, Biochem. J., 2003, 369, 77–88 CrossRef CAS.
- K. Brown, M. Tegoni, M. Prudencio, A. S. Pereira, S. Besson, J. J. Moura, I. Moura and C. Cambillau, Nat. Struct. Biol., 2000, 7, 191–195 CrossRef CAS.
- B. C. Hill, J. Bioenerg. Biomembr., 1993, 25, 115–120 CrossRef CAS.
- D. J. Richardson, L. C. Bell, A. G. McEwan, J. B. Jackson and S. J. Ferguson, Eur. J. Biochem., 1991, 199, 677–683 CAS.
- M. Itoh, K. Matsuura and T. Satoh, FEBS Lett., 1989, 251, 104–108 CrossRef CAS.
- C.-S. Zhang and T. C. Hollocher, Biochim. Biophys. Acta, 1993, 1142, 253–261 CAS.
- I. Matchová and I. Kucera, Biochim. Biophys. Acta, 1991, 1058, 256–260 CAS.
- F. C. Boogerd, H. W. van Verseveld and A. H. Stouthamer, FEBS Lett., 1980, 113, 279–284 CrossRef CAS.
- B. C. Berks, D. Baratta, D. J. Richardson and S. J. Ferguson, Eur. J. Biochem., 1993, 212, 467–476 CAS.
- R. J. Van Spanning, C. Wansell, N. Harms, L. F. Oltmann and A. H. Stouthamer, J. Bacteriol., 1990, 172, 986–996 CAS.
- T. Rasmussen, B. C. Berks, J. Sanders-Loehr, D. M. Dooley, W. G. Zumft and A. J. Thomson, Biochemistry, 2000, 39, 12753–12756 CrossRef CAS.
- P. Chen, S. DeBeer George, I. Cabrito, W. E. Antholine, J. J. Moura, I. Moura, B. Hedman, K. O. Hodgson and E. I. Solomon, J. Am. Chem. Soc., 2002, 124, 744–745 CrossRef CAS.
- V. S. Oganesyan, T. Rasmussen, S. Fairhurst and A. J. Thomson, Dalton Trans., 2004, 996–1002 RSC.
- T. Rasmussen, B. C. Berks, J. N. Butt and A. J. Thomson, Biochem. J., 2002, 364, 807–815 CrossRef CAS.
- J. Riester, W. G. Zumft and P. M. Kroneck, Eur. J. Biochem., 1989, 178, 751–762 CAS.
- J. M. Chan, J. A. Bollinger, C. L. Grewell and D. M. Dooley, J. Am. Chem. Soc., 2004, 126, 3030–3031 CrossRef CAS.
- S. Ghosh, S. I. Gorelsky, P. Chen, I. Cabrito, J. J. Moura, I. Moura and E. I. Solomon, J. Am. Chem. Soc., 2003, 125, 15708–15709 CrossRef CAS.
- J. K. Kristjansson and T. C. Hollocher, J. Biol. Chem., 1980, 255, 704–707 CAS.
- C. L. Coyle, W. G. Zumft, P. M. Kroneck, H. Korner and W. Jakob, Eur. J. Biochem., 1985, 153, 459–467 CrossRef CAS.
- S. W. Snyder and T. C. Hollocher, J. Biol. Chem., 1987, 262, 6515–6525 CAS.
- S. Teraguchi and T. C. Hollocher, J. Biol. Chem., 1989, 264, 1972–1979 CAS.
- K. Sato, A. Okubo and S. Yamazaki, J. Biochem., 1999, 125, 864–868 CAS.
- J. Simon, O. Einsle, P. M. Kroneck and W. G. Zumft, FEBS Lett., 2004, 569, 7–12 CrossRef CAS.
- C. D. Richter, J. W. Allen, C. W. Higham, A. Koppenhofer, R. S. Zajicek, N. J. Watmough and S. J. Ferguson, J. Biol. Chem., 2002, 277, 3093–3100 CrossRef CAS.
- D. L. Brautigan, S. Ferguson-Miller and E. Margoliash, Methods Enzymol., 1978, 53, 128–164 CAS.
- P. Mendes, Comput. Appl. Biosci., 1993, 9, 563–571 CAS.
- M. Prudencio, A. S. Pereira, P. Tavares, S. Besson, I. Cabrito, K. Brown, B. Samyn, B. Devreese, J. Van Beeumen, F. Rusnak, G. Fauque, J. J. Moura, M. Tegoni, C. Cambillau and I. Moura, Biochemistry, 2000, 39, 3899–3907 CrossRef CAS.
- L. M. Geren, J. R. Beasley, B. R. Fine, A. J. Saunders, S. Hibdon, G. J. Pielak, B. Durham and F. Millett, J. Biol. Chem., 1995, 270, 2466–2472 CrossRef CAS.
- K. Wang, Y. Zhen, R. Sadoski, S. Grinnell, L. Geren, S. Ferguson-Miller, B. Durham and F. Millett, J. Biol. Chem., 1999, 274, 38042–38050 CrossRef CAS.
Footnote |
| † Based on the presentation given at Dalton Discussion No. 8, 7–9th September 2005, University of Nottingham, UK. |
| This journal is © The Royal Society of Chemistry 2005 |