Synthesis, characterization, in vitro antitumor activity, DNA-binding properties and electronic structure (DFT) of the new complex cis-(Cl,Cl)[RuIICl2(NO+)(terpy)]Cl†
Konstantina Karidiab, Achilleas Garoufisa, Athanassios Tsipisa, Nick Hadjiliadisa, Hans den Dulkb and Jan Reedijk*b
aLaboratory of Inorganic and General Chemistry, Department of Chemistry, University of Ioannina, Ioannina 45110, Greece. E-mail: nhadjis@cc.uoi.gr; Fax: +30 26510 44831; Tel: +30 26510 98420agaroufi@cc.uoi.gr; Tel: + 30 26510 98409
bLeiden Institute of Chemistry, Gorlaeus Laboratories, Leiden University, 2300 RA Leiden, The Netherlands. E-mail: reedijk@chem.leidenuniv.nl; Fax: 31 71 527 4671; Tel: 31 71 527 4459
First published on 18th February 2005
Abstract
The complex cis-(Cl,Cl)-[RuCl2(terpy)(NO)]Cl (1) has been synthesized by the reaction of [RuCl3(H2O)2(NO)] with terpyridine (terpy) and characterized by various spectroscopic, analytical techniques and using electronic structure calculation (DFT) methods. The cytotoxic activity and the DNA-binding properties of 1 have also been studied using biochemical techniques. The results establish unequivocally that 1 corresponds to a so-called [RuNO]6 species, which readily releases the nitrosyl ligand upon irradiation with a mercury lamp in acetonitrile solution. DFT calculations provided a satisfactory description of structural, bonding, electronic and related properties of the new compound and throw light on the mechanism of the photo-induced NO release. Screening on A2780 (human ovarian carcinoma) cell lines showed significant cytotoxicity with an IC50 value of 0.49 µM. 31P and 23Na NMR spectroscopy along with electrophoretic mobility studies illustrated that complex 1 primarily binds by coordination to DNA without any π-interaction between the planar terpy ligand and the DNA bases, while weak electrostatic interactions could not be excluded. Studies on the inhibition of the restriction enzymes DraI and SmaI revealed that 1 prefers the guanine and cytosine bases of DNA.
1 Introduction
The discovery that nitric oxide plays an important role in many biological processes1 has stimulated enormous interest in the chemistry and biochemistry of NO. Nitric oxide has been shown to be of key importance in many bioregulatory functions, including cardiovascular control,2 neuronal signaling3 and as an agent for defense mechanisms against microorganisms and tumors.4 Behind these multiple functions, an enzyme, the NO synthase (NOS) is responsible for the production of NO in the cell.5 The regulation of nitric oxide in the organism has led to the development of drugs, which either release nitric oxide, or act as NO scavengers.Compounds that liberate nitric oxide under light irradiation, have attracted considerable interest.6 Among them, nitrosyl ruthenium complexes have been proposed as attractive therapeutic agents in biomedicine7,8 and in tumor phototherapy (PDT).9–13 A number of nitrosyl ruthenium complexes containing the polypyridine ligands 2,2′-bipyridine (bpy) or 2,2′:6′, 2″-terpyridine (terpy) have been synthesized and extensively studied over the last few decades, and only a selection of recent references is given here.14–23 Meyer et al.22–25 have synthesized and characterized a number of nitrosyl ruthenium complexes with imine N-donor co-ligands. Interestingly, the effect of the photo-induced NO release from the cis-[RuCl(NO)(bpy)2)](PF6)2 complex in aqueous media has been studied by flash-photolysis at 355 nm light,16 while laser pulse photolysis has been used to control NO liberation in a series of nitrosylruthenium complexes formulated as cis-[Ru(NO)(bpy)2L]3+ (L = py, 4-picoline, 4-acetylpyridine).14 Also the mixed-ligand chloro-polypyridyl ruthenium complexes [RuCl(terpy)(bpy)]Cl, cis-[RuCl2(bpy)2], and mer-[Ru(terpy)Cl3] (terpy = 2,2′:6′,2″-terpyridine, bpy = 2,2′-bipyridyl) were screened for antitumor activity against murine and human tumors and show favourable antitumor properties.26 More particularly, the complex mer-[Ru(terpy)Cl3], was found to be a relevant cytotoxic compound, able to bind to DNA firmly and to modify importantly its conformation.26 In this study, it was also confirmed that mer-[Ru(terpy)Cl3] unwinds DNA and coordinates preferentially to isolated guanine bases. The reaction between the complex cis-[Ru(bpy)2Cl2] and 9-EtGua in 1 : 1 molar ratio has been reported, and the crystal structure of the [RuCl(bpy)2(9-EtGua)]Cl complex formed revealed that the 9-EtGua coordinates through its N7 atom.27 Very recently an X-ray structure of a bis(9-meguanosine) adduct of Ru(bpy)2 has been reported.28
The unique applications of light and nitrosylruthenium complexes create a real challenge for the development of improved antitumor compounds.29 The aim of our research efforts is the discovery of novel antitumor agents combining the cytotoxic activity of chloro-polypyridyl ruthenium complexes with the photoreactivity of nitrosylruthenium complexes, as potential therapeutic agents in photodynamic therapy (PDT). The compound trans-(Cl,Cl)-[RuIICl2(terpy)(NO+)]Cl has been reported19 and discussed, but the corresponding cis isomer is structurally unknown. In this paper we present the synthesis, the characterization, the cytotoxic activity and the DNA-binding properties of the new complex cis-(Cl,Cl)-[RuIICl2(terpy)(NO+)]Cl (1), which exhibits remarkable cytotoxic activity. Electronic structure calculations based on density functional theory (DFT) methods have also been used to describe the structural, bonding, electronic and related properties of the new complex and unravel the mechanism of the photo-induced NO release process from the complex under light irradiation.
2 Experimental
2.1 Instrumentation
The infrared spectra of the complexes in the 4000–300 cm−1 range were recorded on a Perkin-Elmer Paragon 1000 FTIR spectrophotometer equipped with a Golden Gate Diamond ATR device, using the diffuse reflectance technique. C, H and N determinations were performed on a Perkin-Elmer 2400 Series II analyzer. UV-Visible spectra were recorded on a Varian Cary 3-Bio spectrophotometer with temperature controller. The irradiation of the solutions was carried out with a Philips mercury lamp. Thermal denaturation experiments were performed in quartz cuvettes. Samples were continuously heated with a 1 °C min−1 rate of temperature increase, while monitoring the absorbance changes at 260 nm. The investigated interval of temperature ranged from 24 to 98 °C. Values for melting temperatures (Tm) and for the melting interval (ΔT) were determined according to the reported procedures.30 CD spectra were recorded on a Jobin Yvon CD-6 instrument at room temperature. The LC-MS experiments were performed on a Finnigan MAT TSQ-700 instrument with a custom-made electrospray interface combined with LC-equipment consisting of a Dionex pump P580, a Gilson 119 UV-detector at 214 and 254 nm and an Alltima C18 150 mm × 4.6 mm ID column. For the electrophoretic mobility assay the samples were analyzed by gel electrophoresis on 0.8% (w/v) agarose gel at 10 V cm−1. The optical density for the cytotoxicity experiment was measured by microplate reader (Bio Rad) at 590 nm. IC50 values were obtained by GraphPad Prism software, version 3.05, 2000. The 23Na NMR and 31P NMR spectra were obtained on a Bruker 300 DPX spectrometer operating at 121.49 MHz and at 79.39 MHz for 31P and 23Na NMR, respectively at 310 K in D2O solutions. The DNA concentration was 6 mM and the complex concentration in the solution was varied from 0.6 to 3 mM.2.2 Materials
2,2′:6′,2″-Terpyridine was purchased from Aldrich Chemical Company and used without further purification. 9-Ethylguanine, calf thymus DNA, agarose, ethidium bromide and PIPES (piperazine N,N′-bis(2-ethanesulfonic acid)) were purchased from Sigma Chemical Company. Hydrated ruthenium trichloride, RuCl3·3H2O, was obtained as a loan from Johnson Matthey. The [RuCl3(NO)(H2O)2] complex was prepared according to a literature procedure.31 Plasmid pUC9, 2665bp, was isolated and purified twice with CsCl gradient centrifugation. The plasmid isolation contained over 95% of supercoiled DNA with a faint relaxed DNA band. The restriction enzymes DraI (three recognition sites of TTTAAA at position 1073,1765 and 1784 and SmaI (recognition site GGGCCC at position 260) were used in this work for the digestion of the supercoiled DNA. The sonication of the calf thymus DNA was performed according to the literature.322.3 Preparation of ruthenium adducts with DNA for CD spectra
The reactions between the cis-(Cl,Cl)-[RuCl2(NO)(terpy)]Cl (1) complex and calf thymus DNA were carried out by adding the required volume of a freshly prepared solution of the complex dissolved in 1 mM PIPES and 20 mM sodium chloride aqueous solution, to calf thymus DNA solutions (100 µM) and incubating at 25 °C for 24 h. Samples were prepared in such a way as to have final ruthenium/DNA base pair ratios (r) of 0.1, 0.3, and 0.5 (complex concentration was 10, 30 and 50 µM). Electronic and CD spectra were recorded at room temperature. The DNA concentration, expressed as moles of nucleotides per liter, [P], was determined from the absorbance at 260 nm (ε260 = 6600 M−1 × cm−1, T = 298 K). The sample displayed a value of the A260/A280 ratio of 1.9, consistent with low protein content.2.4 Preparation of ruthenium adducts with DNA for electrophoretic mobility assays
Adducts with pUC9 plasmid DNA were prepared by adding the required volume of a freshly prepared solution of the complex cis-(Cl,Cl)-[RuCl2(NO)(terpy)]Cl (1) using 10 mM PIPES and 4 mM sodium chloride aqueous solution. The concentration of pUC9 DNA in the reaction mixture was 38 ng ml−1, while the concentration of the complex was varied to give different metal-to-base pair stoichiometries (0.1, 0.3, 0.5, 0.7, and 1). The supercoiled plasmid DNA was incubated for 2 h at RT and each sample was purified using GFX DNA and Gel Band Purification Kit (Amersham 27-9602-01) to remove excess metal complex not bound to DNA. The mobility of the complex-treated pUC9 samples was analyzed by agarose gel electrophoresis at RT for 2 h in Tris-acetate/edta buffer, and then the gel was stained for 1 h in 0.5 mg ml−1 (w/v) ethidium bromide.2.5 Digestion of plasmid DNA by restriction enzymes
Enzyme digestions were carried out by incubating the untreated and cis-(Cl,Cl)-[RuCl2(NO)(terpy)]Cl-treated pUC9 samples (at r = 0.1, 0.3) with DraI and SmaI, respectively. 200 ng of each sample were incubated with the restriction enzymes at 37 °C for 1 h in the appropriate buffer recommended by the manufacturer. Then DNA restriction fragments were analysed by agarose gel electrophoresis 0.8% (w/v) and the gel was stained for 1 h in 0.5 mg ml−1 (w/v) ethidium bromide.2.6 Generation of photoproducts for FTIR analysis
The complex cis-(Cl,Cl)-[RuCl2(NO)(terpy)]Cl (1) was dissolved in degassed acetonitrile. The brown solution was irradiated with light from a mercury lamp, for 2 h until the color of the solution became orange. The solvent was removed from the mixture by a stream of argon leaving a solid orange residue, which was analyzed by infrared spectroscopy.2.7 Generation of photoproducts for UV-visible analysis
The complex cis-(Cl,Cl)-[RuCl2(NO)(terpy)]Cl (1) was dissolved in degassed acetonitrile in a concentration of 10−4 mol l−1, and the solution was placed in a quartz cuvette with 1 cm path length. The solution was irradiated with a mercury lamp.2.8 Cell lines and cytotoxicity test
A2780 (human ovarian carcinoma) and A2780 cisplatin-resistant cell lines were maintained in continuous logarithmic culture in Dulbecco's modified Eagle's medium (DMEM) (Gibco BRL, Invitrogen Corporation, The Netherlands) supplemented with 10% fetal calf serum (Perbio Science, Belgium), penicillin G sodium (100 units ml−1, Duchefa Biochemie BV, The Netherlands), streptomycin (100 µg ml−1 Duchefa Biochemie BV, The Netherlands), and Glutammax 100× (Gibco BRL, The Netherlands). The cells were harvested from confluent monolayers.For the cytotoxicity evaluation, 2000 cells well−1 were seeded in 100 µl of complete medium in 96-multiwell flat-bottom microtiter plates (Corning Costar). The plates were incubated at 37 °C in 5% CO2 for 48 h prior to drug testing to allow cell adhesion. The stock solutions (1 mg ml−1 DMSO) of the tested compound were freshly prepared and directly used for the dilutions. Growth inhibition by the complex cis-(Cl,Cl)-[RuCl2(NO)(terpy)]Cl (1) was determined using an MTT-based assay33 using a previously described method.34 In the cytotoxicity test, a DMSO stock solution was used due to the poor solubility of the complex in water (∼3 × 10−3 M at 310 K) in order to have a better comparison with other cytotoxic compounds (see Section 3.3 below). The cells had been exposed to the compound for a relatively long time (3 days), to allow the compound to be able to reach DNA, or another biological target, and act on this level. The IC50 values represent the concentration of a drug that is required for 50% reduction of cellular growth.
2.9 Preparation of [Ru(terpy)NOCl2]Cl (1)
[RuCl3(H2O)2(NO)] (0.1 g, 0.365 mmol) and terpy (0.085 g, 0.365 mmol) were suspended in EtOH (20 ml) and stirred overnight at room temperature. A brown solid was precipitated and the solution was filtered. The filtrate was left at room temperature and after some time (3 days) dark brown crystals were precipitated. The product was collected by filtration, washed with diethyl ether (2 × 5 ml) and dried in vacuo. (75 mg, 0.16 mmol, Yield 44%) RuC15H11N4OCl3 (MW 470.5).Anal. Calcd. for RuC15H11N4OCl3: C, 38.28; H, 2.36; N, 11.90%, Found: C, 38.34; H, 1.98; N, 11.58). ESI-MS: m/z 435, [RuCl2(terpy)(NO)]+. IR ν(NO) = 1860 cm−1 (s). 1H NMR (300 MHz, DMSO-d6): δ 9.15 (d, 3JHH = 5.0 Hz, 2H, H6H6″), 8.94 (t, 3JHH = 7.8 Hz, 4H, H3′H5′, H3H3″), 8.83 (t, 3JHH = 7.2 Hz, 1H, H4′), 8.50 (t, 3JHH = 7.8 Hz, 2H, H4H4″), 7.98 (t, 3JHH = 6.8 Hz, 2H H5,5″). The low value for the %H (0.38%) is ascribed to experimental error.
2.10 Computational details
The DFT calculations at the B3LYP level of theory on the ruthenium complexes were studied using the GAUSSIAN03 program suite.35 The geometries of all species were fully optimized at the Becke's 3-Parameter hybrid functional36,37 combined with the Lee–Yang–Parr38 correlation functional termed as B3LYP level of density functional theory. Three basis sets have been used for calculations including the SDD basis set which describes valence electrons with a [8s,7p,6d/6s,5p,3d] valence basis set and Stuttgart–Dresden relativistic ECP's on the ruthenium atom,39 the SDD basis set for the ruthenium atom and the larger all-electron double-zeta (DZVP) basis set40 for the non-metal atoms; the LANL2DZ basis set which describes valence electrons with a [5s,6p,4d/3s,3p,2d] valence basis set and Los Alamos ECP's on the ruthenium atom41,42 and the 6-31G(d,p) basis for the non metal atoms. Full geometry optimization was performed for each structure using Schlegel's analytical gradient method43 and the attainment of the energy minimum was verified by calculating the vibrational frequencies that result in absence of imaginary eigenvalues. All the stationary points have been identified for minimum (number of imaginary frequencies NIMAG = 0) or transition states (NIMAG = 1). The vibrational modes and the corresponding frequencies are based on a harmonic force field. The computed electronic energies, were corrected to constant pressure and 298 K, for zero point energy (ZPE) differences and for the contributions of the translational, rotational and vibrational partition functions. The natural bond orbital (NBO) population analysis was performed using Weinhold's methodology.44 Time-dependent density functional theory (TD-DFT)45–47 calculations were performed on the equilibrium ground state geometries employing the same density functionals and basis sets used in geometry optimization. The Davidson algorithm was used, in which the error tolerance in the square of the excitation energies and trial-vector orthonormality criterion were set to 10−8 and 10−10, respectively. The success of TD-DFT method in calculating excitation energies of transition metal complexes has been demonstrated in several recent studies.48,493 Results and discussion
3.1 Synthesis, characterization and photochemistry of the complex cis-(Cl,Cl)-[RuCl2(NO)(terpy)]Cl (1)
When the complex anion [RuCl5(NO)]2− reacts with terpy it yields the complex trans-(Cl,Cl)-[RuCl2(NO)(terpy)]+ (2), which has been structurally characterized earlier.19 In this reaction KCl was added to prevent the fast hydrolysis of chloride in the parent complex, as the chloride ligand in a trans position to NO ligand can easily be replaced by a solvent molecule.50 Thus, the terpy ligand is coordinated to the ruthenium ion in a tridentate bonding fashion leaving the remaining chloride ligands in trans positions to each other. The characteristic ν(NO) stretching vibration band appears at 1895 cm−1 in (2). In the same reaction of [RuCl5(NO)]2− with terpy, a minor product is also formed, which could be either a geometric isomer or a nitrosyl complex containing H2O, and/or OH,− instead of Cl− and exhibiting a ν(NO) stretching frequency at higher wavenumbers (1920 cm−1). On the other hand, the reaction of terpy with the [RuCl3(NO)(H2O)2] complex prepared by the method of Fletcher et al.,31 afforded a new complex, with a ν(NO) stretching vibration at 1860 cm−1. The elemental analysis revealed that both water molecules in the resulting complex were replaced by the pyridine rings of terpy and that the new complex could be formulated as [RuCl2(NO)(terpy)]Cl (1). Considering that the ν(NO) stretching vibration band of 1 was shifted to lower wavenumbers by about 35 cm−1 with respect to that of the trans counterpart, 2, we have to assume that the new complex corresponds to the (other) cis-geometric isomer of the complex [RuCl2(NO)(terpy)]Cl; (See Scheme 1).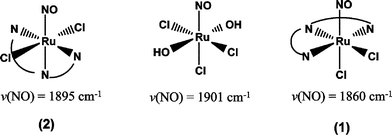 | ||
| Scheme 1 Ruthenium NO complexes and reaction products with terpy, with NO stretching frequencies. | ||
The frequency of the ν(NO) stretching vibration in nitrosylruthenium complexes has been suggested to be a good indicator of the relative coordination site of the NO ligand with respect to the coordination sites of the nitrogen donor atoms of terpy or the chloride ligands. Thus, in the [RuCl5(NO)]2− complex, which involves a coordinated NO ligand in a trans position to a chloride ligand, the ν(NO) stretching vibration absorbs at 1904 cm−1.51 The [RuCl3(NO)(H2O)2]Cl complex exhibiting a ν(NO) stretching vibration at 1901 cm−1 should also involve a coordinated NO ligand in a trans position to a chloride ligand. On the other hand, for a coordinated NO ligand in a trans position to a nitrogen donor atom of a pyridine ring, the ν(NO) stretching vibration is known to be shifted towards higher frequencies (e.g.cis-[Ru(NO)(bpy)2Cl](PF6)2, ν(NO) = 1931 cm−1,52cis-[Ru(NO)(bpy)2(py)](PF6)3, ν(NO) = 1950 cm−1.52 Interestingly, the ν(NO) stretching vibration of the trans-[RuCl(NO)(bpy)2](PF6)2 complex absorbing at 1912 cm−1 matches those of the [RuCl5NO]2− and [RuCl3(NO)(H2O)2]+ complexes involving the nitrosyl ligand in a trans position to a chloride ligand.53 This effect could probably be a consequence of the offsetting between the trans-strengthening effect of the chloride ligands (σ/π-donor ligands) and the modest cis weakening effect due to the four nitrogen donor atoms of the bpy ligands (σ-donor/π-acceptor ligands). Taking into account that in the trans-(Cl,Cl)-[RuCl2(NO)(terpy)]+ complex (2), involving the NO ligand in a trans position to a pyridine ring of terpy, the ν(NO) stretching vibration absorbs at 1895 cm−1, the isolated new complex with a ν(NO) stretching vibration at 1860 cm−1 should therefore correspond to the geometric isomer, cis-(Cl,Cl)-[RuCl2(NO)(terpy)]Cl (1).
The purity of the complex was checked by LC-MS spectrometry using acetonitrile as a carrier solvent and a clear positive ion ESI-MS spectrum was obtained (Fig. 1). The peak centered at m/z = 435 corresponds to the single charged cation [RuCl2(NO)(terpy)]+ as confirmed from the calculated isotopic pattern.
![The ESI-MS positive ion spectrum from the complex [RuCl2(NO)(terpy)]Cl. Inset: The calculated spectrum for the cation [RuCl2(NO)(terpy)]+. Units m/z in Da.](/image/article/2005/DT/b418838a/b418838a-f1.gif) | ||
| Fig. 1 The ESI-MS positive ion spectrum from the complex [RuCl2(NO)(terpy)]Cl. Inset: The calculated spectrum for the cation [RuCl2(NO)(terpy)]+. Units m/z in Da. | ||
In the electronic spectrum (Fig. 2, t = 0 min) of the cis-(Cl,Cl)-[RuCl2(NO)(terpy)]Cl complex in acetonitrile solution (10−4 M) the intense band at 274 nm is most likely the same π → π*(terpy) transition that is commonly found in this region for typical polypyridine complexes.23,54 The bands at 330 and 349 nm are possibly MLCT bands (dπ(Ru) → π*(NO) and dπ(Ru) → π*(terpy).23,24 During the irradiation the light orange color of the solution turned to deep orange–brown accompanied by changes in the UV-Vis spectrum. A decrease in the absorption bands at 330 and 349 nm and an increase in absorptions at 309 and 414 nm were observed. Exhaustive irradiation leads to a final stable spectrum. Similar changes were reported for the cis-[RuCl(NO)](bpy)22+] complex,14,16 the cis-[Ru(NO)(PaPy3)]2+ complex (PaPy3H = N,N′-bis(2-pyridylmethyl)amine-N-ethyl-2-pyridine-2-carboxamide)55 and the [RuCl(salen)(NO)] complex (salen = N,N′-ethylenebis(salicylideneiminato)dianion),56 indicating the photorelease of the NO ligand. The presence of isosbestic points in the successive spectra of the irradiated solution, suggests the transformation of the complex to only one final photoproduct (Fig. 2).
![Electronic spectra of the cis-(Cl-Cl)-[RuCl2(terpy)(NO)]Cl complex in CH3CN during light irradiation.](/image/article/2005/DT/b418838a/b418838a-f2.gif) | ||
| Fig. 2 Electronic spectra of the cis-(Cl-Cl)-[RuCl2(terpy)(NO)]Cl complex in CH3CN during light irradiation. | ||
In the IR spectrum of the irradiated solution the absence of any band corresponding to the NO stretching vibration indicates its dissociation from the ruthenium complex. Additionally, the EPR spectrum of the photoproduct showed paramagnetic species suggesting the presence of Ru(III). The LC-MS analysis of the photoproduct indicated the presence of only one complex corresponding to the solvated complex cis-(Cl,Cl)-[RuCl2(terpy)(CH3CN)]Cl. However, the ESI-mass spectrum shows a peak at m/z 404.9 assigned to the [Ru(terpy)Cl2]+ cationic species which is in accordance with the calculated spectrum (Fig. 3). The dissociation of the solvent molecule in the gas phase during the ESI process (collision energy 80 eV), probably takes place due to the weak bonding between Ru(III) and the acetonitrile. The overall photochemical reaction can be represented by the following chemical equation:

![The ESI-MS positive ion spectrum of the cation [RuCl2(terpy)]+.](/image/article/2005/DT/b418838a/b418838a-f3.gif) | ||
| Fig. 3 The ESI-MS positive ion spectrum of the cation [RuCl2(terpy)]+. | ||
3.2 Computational studies using DFT
Comparison of the DFT geometry optimization results with the available experimental X-ray data for the trans isomer show that the bond lengths and angles in the trans-(Cl,Cl)-[RuCl2(NO)(terpy)]+ complex are reproduced quite well for all basis sets used. The salient feature of the equilibrium geometries of the two isomers is the shortening of the Ru–Cl bond in the trans position to the NO ligand by 4.3 pm. This is in agreement with the trans-shortening effect. The coordination polyhedron around ruthenium in 1 is a slightly distorted octahedron with Cs symmetry, the mirror plane including the Ru, N(1), N(3), Cl(1) and Cl(2) atoms. Moreover, the nitrogen donor atoms of terpy and the Cl(1) atom define an equatorial plane with the Cl(2) and the NO ligands in the axial positions. The Ru atom is displaced 16.3 pm toward the nitrosyl ligand from the equatorial plane. Such a displacement is a characteristic feature of nitrosylruthenium complexes; for example the Ru atom is displaced by 12.8 pm towards the nitrosyl ligand in the[RuCl(salen)(NO)] complex56 and 18.8 pm in the [Ru(CN)5(NO)]2− complex.57 Overall, the equilibrium structure of 1 is consistent with an almost linear Ru–N–O bonding scheme, typical for [NO]+ coordinating to a d6 RuII metal center.58
Surprisingly, most of the geometrical parameters are reproduced better using the smaller SDD basis set. Even with the SDD basis set the unscaled ν(NO) harmonic vibrational frequency of 1829 cm−1 is significantly closer to the experimental value of 1895 cm−1 (3.5% lower) for complex 2 than those of 1993 and 1987 cm−1 (∼5% higher) computed with the larger LANL2DZ(Ru)-6-31G(d) and SDD(Ru)-DZVP basis sets, respectively. These deviations of the computed ν(NO) harmonic vibrational frequencies are found at the accuracy limits (±5%) for the vibrational frequencies calculated by the most rigorous quantum chemical computational methods. Generally, the computed bond lengths are slightly overestimated for all basis sets used. This is not an unexpected result, since the calculations were performed on the complexes in a vacuum (gas phase).
The unscaled ν(NO) harmonic vibrational frequency of 1812 cm−1 of the new complex cis-(Cl,Cl)-[RuCl2(NO)(terpy)]+ computed with the smaller SDD basis set deviates from the experimental value of 1860 cm−1 by only 2.6%, while using the larger basis sets the deviations are much larger (6.7% and 5.8%) with the ν(NO) bands being shifted to higher frequencies. It should be noted that in the [RuCl(salen)(NO)] complex,56 involving the NO ligand in a trans position to a chloride ligand, the ν(NO) band occurs at 1838 cm−1. Our calculations correctly predicted that the ν(NO) stretching vibration of the cis isomer absorbs at lower frequencies (by about 17 cm−1 if the SDD basis set is used) than the trans isomer in line with the experimental observations. Therefore, we can expect that for nitrosylruthenium complexes the structural and spectroscopic properties can be “pin-pointed” by the B3LYP functional using the relatively small SDD basis set.
According to the Natural Bond Orbital (NBO) and Mulliken population analysis (Table 1), the N–O bond in 1 acquires a bond overlap population (bop) of 0.314 which is found between the bop values of the N–O bond in the free [NO]˙ (0.217) and [NO]+ (0.410) ligands. Obviously, upon coordination the [NO]+ ligand loses some of the triple bond character. Upon coordination about 0.25 charge units of natural charge is transferred from the [NO]+ ligand to the d6 RuII central atom of the cis-(Cl,Cl)-[RuCl2(terpy)] fragment in the 2A ground state, while concomitantly both the N-donor and O atoms accept a charge density of about 0.47 and 0.31 charge units of natural charge, respectively. These electron charge-transfer processes are indicative of a strong π-back donation from the Ru(II) to [NO]+ ligand. Notice the very small lowering of the positive natural atomic charge on the Ru(II) central atom by only 0.05 charge units upon coordination of the [NO]+ ligand, which is the result of a balance between the σ-donation and π-back donation characterizing the RuII–(NO)+ bond. The balance between the two opposite charge transfer processes is also mirrored on the natural electron configuration of the Ru(II) central atom (Table 1), which remains almost unchanged upon the coordination of the [NO]+ ligand. Both charge transfer processes strengthen the Ru–NO and weaken the N–O bonds. Consequently, the calculated Ru–N bond length is short and the N–O bond length is significantly longer than the expected value for a free [NO]+. The N–O bond in the [NO]+ molecule has a predicted bond length of 110.7 pm at the B3LYP/SDD level. The relatively strong RuII–(NO)+ interaction is also reflected in the computed high binding energy of the [NO]+ ligand to the [RuIICl2(terpy)] fragment amounting to about 148 kcal mol−1 at the B3LYP/SDD level of theory. The binding energy of the [NO]˙ (S = 1/2) ligand to [RuIIICl2(terpy)]+ fragment is much lower, and amounts to only 48 kcal mol−1. This behaviour is expected on the grounds of the reduced π back-donation in the [RuIIICl2(terpy)(NO)˙]+ complex, as well as to the addition of an extra electron into a π* MO of the coordinated NO ligand. This bonding mechanism characterises the light-induced excited triplet state of the complex. Finally, the high binding energy of the [NO]− ligand to the [RuIVCl2(terpy)]2+ fragment amounting to about 330 kcal mol−1, is the result of the strong electrostatic interactions between the negatively charged [NO]− ligand and the Ru(IV) central atom.
| Natural electron configuration | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Ru | N(1) | O | |||||||||
| Complex | qRu | qN(1) | qO | bop (N–O) | 5s | 4d | 5p | 2s | 2p | 2s | 2p |
| cis-[RuCl2(NO)(terpy)]+ | 0.59 | 0.31 | −0.10 | 0.314 | 0.36 | 6.97 | 0.01 | 1.44 | 3.20 | 1.75 | 4.33 |
| cis-[RuCl2(terpy)] | 0.64 | 0.30 | 7.02 | 0.01 | |||||||
| cis-[RuCl2(terpy)]+ | 0.85 | 0.34 | 6.76 | 0.02 | |||||||
| cis-[RuCl2(terpy)]2+ | 0.90 | 0.35 | 6.71 | 0.01 | |||||||
| [NO]+ | 0.79 | 0.21 | 0.410 | 1.73 | 2.46 | 1.77 | 4.00 | ||||
| [NO]˙ | 0.20 | −0.20 | 0.217 | 1.76 | 3.01 | 1.79 | 4.40 | ||||
| [NO]− | −0.43 | −0.57 | 0.054 | 1.80 | 3.59 | 1.82 | 4.74 | ||||
The [NO]+ ligand is bound to the Ru(II) via a donor σ bond, comprising of the N lone pair delocalizing into an empty metal σ orbital and a π back bonding consisting of a combination of the filled dxy and dyz orbitals of the Ru(II) central atom and the doubly degenerate, empty π* orbitals of the [NO]+ ligand. The bonding σ(Ru–NO) interaction between the Ru and N donor atom of the NO ligand in 1 is constructed from the 4dxz natural orbital on the Ru atom interacting with the 2px natural orbital on the N atom, thus having the form σ(Ru–NO)cis = 0.8089(4dxz)Ru + 0.5879(2px)N.
| λ/nm | fa | Excitation | Composition of the participating MOs |
|---|---|---|---|
| a Only transitions with oscillator strengths stronger than 0.001 are considered. | |||
| 411 | 0.0231 | HOMO − 1 → LUMO | (dπRu − 3pCl) → (dπRu − π*-NO) |
| 410 | 0.0380 | HOMO − 2 → LUMO + 1 | (dπRu + π*-NO − 3pCl) → (dπRu − π*-NO) |
| 404 | 0.0410 | HOMO − 3 → LUMO + 1 | (dπRu + π*-NO − 3pCl) → (dπRu − π*-NO) |
| 400 | 0.0540 | HOMO − 3 → LUMO | (dπRu + π*-NO − 3pCl) → (dπRu − π*-NO) |
| 352 | 0.0057 | HOMO → LUMO + 5 | (dπRu − 3pCl) → (π*-terpy) |
| 351 | 0.0293 | HOMO − 4 → LUMO + 1 | (π-terpy) → (dπRu − π*-NO) |
| 330 | 0.0171 | HOMO − 5 → LUMO + 1 | (dπRu + π*-NO + 3pCl + π-terpy) → (dπRu − π*-NO) |
| 328 | 0.0426 | HOMO − 5 → LUMO + 1 | (dπRu + π*-NO + 3pCl + π-terpy) → (dπRu − π*-NO) |
| 323 | 0.1533 | HOMO − 1 → LUMO + 1 | (dπRu − 3pCl) → (dπRu − π*-NO) |
| 318 | 0.0064 | HOMO − 6 → LUMO + 1 | (dπRu + π*-NO + 3pCl + π-terpy) → (dπRu − π*-NO) |
It can be seen from Table 2 that the computed electronic transitions of the cis-(Cl,Cl)-[RuCl2(NO)(terpy)]+ complex are in good agreement with the experimental ones discussed in the text (experimental values are 330 and 349, theoretical values are 323 and 352 nm). However, because of the highly delocalized MOs involved in the electronic transitions and the high mixing of excited configurations due to the low symmetry of the complexes, it is very difficult to assign the calculated transitions into the well-known intraligand (LL), ligand-to-metal and metal-to-ligand charge transfer (LMCT and MLCT) and ligand field (d → d) transitions. The band with the higher intensity in 1 is predicted to occur at 323 nm, in excellent agreement with experiment. This band arises from the electronic transitions from the HOMO → LUMO + 4 and HOMO − 1 → LUMO + 1, as a result of the mixing of excited configurations having large coefficients in the CI wave functions. To intuitively understand the absorption processes the density diagrams of the participating MOs are depicted in Fig. 4.
![Single-electron transitions with the CI coefficients in the TD-DFT calculations for the more intense band of the cis-(Cl,Cl)-[RuCl2(terpy)(NO)]Cl complex.](/image/article/2005/DT/b418838a/b418838a-f4.gif) | ||
| Fig. 4 Single-electron transitions with the CI coefficients in the TD-DFT calculations for the more intense band of the cis-(Cl,Cl)-[RuCl2(terpy)(NO)]Cl complex. | ||
It can be seen from Fig. 4 that this band could be classified as a charge transfer (CT) band resulting from a combination of dπ(Ru) → π*(terpy) and dπ(Ru) → π*(NO) excitations. The band absorbing at 351 nm (experimental value 349 nm) corresponds to an intraligand π(terpy) → π*(NO) transition. Moreover, the transitions absorbing in the region of 400 to 411 nm must be primarily MLCT transitions.
| MSII | MSI | 4 | 5 | ||||
|---|---|---|---|---|---|---|---|
| Parameter | 3 (S = 3) | (S = 1) | (S = 3) | (S = 1) | (S = 3) | ||
| Re(N1–O) | 120.7 | 125.5 | 126.7 | 118.7 | 121.0 | ||
| Re(Ru–N1) | 193.9 | 195.1 | 224.1 | 206.2 | |||
| Re(Ru–O) | 221.2 | 221.3 | 187.1 | 214.6 | |||
| Re(Ru–Cl1) | 235.8 | 245.7 | 239.0 | 242.7 | 236.8 | 235.3 | 239.0 |
| Re(Ru–Cl2) | 241.1 | 238.2 | 235.5 | 234.8 | 235.9 | 233.7 | 238.8 |
| Re(Ru–N2) | 210.0 | 209.3 | 208.9 | 209.6 | 209.1 | 208.3 | 208.9 |
| Re(Ru–N3) | 204.3 | 202.1 | 203.2 | 200.5 | 202.6 | 201.5 | 201.8 |
| ∠Ru–N1–O | 140.0 | 84.3 | 72.3 | ||||
| ∠Ru–O–N1 | 171.0 | 135.9 | |||||
| ∠N1–Ru–Cl1 | 89.2 | 76.0 | 77.0 | 85.4 | |||
| ∠O–Ru–Cl1 | 85.6 | 86.1 | |||||
| ∠N1–Ru–Cl2 | 180.0 | 158.2 | 161.8 | 176.4 | |||
| ∠O–Ru–Cl2 | 175.9 | 177.5 | |||||
| ∠Cl1–Ru–Cl2 | 90.7 | 82.1 | 84.8 | 90.3 | 91.9 | 103.0 | 91.0 |
| ∠N1–Ru–N2 | 91.5 | 96.5 | 91.9 | 90.1 | |||
| ∠O–Ru–N2 | 93.4 | 87.4 | |||||
![The energetic and geometric profile of the photo-induced NO release from cis-(Cl,Cl)-[RuCl2(terpy)(NO)]Cl complex computed at the B3LYP/SDD level.](/image/article/2005/DT/b418838a/b418838a-f5.gif) | ||
| Fig. 5 The energetic and geometric profile of the photo-induced NO release from cis-(Cl,Cl)-[RuCl2(terpy)(NO)]Cl complex computed at the B3LYP/SDD level. | ||
Upon light irradiation 1 is initially excited to the triplet state 3, which subsequently releases the NO molecule, either directly or through the metastable states MSII and MSI, yielding the final solvated species 5. The existence of the photoirradiation-induced long-lived metastable states MSI and MSII in nitrosyl complexes has been confirmed both experimentally and theoretically for nitrosylruthenium complexes.14,61–63 The equilibrium structures of the metastable states were calculated starting from the model proposed by Carducci et al.,63 which proposes an O-end-on (η1-ON) bonding mode for MSI and a side-on (η2-NO) bonding mode for MSII. Both MSI and MSII are diamagnetic species corresponding to local minima on the ground state potential energy surface. The excited triplet state and the two metastable states are connected with a geometrical and electronic rearrangement of the coordinated NO ligand and are energetically lifted by 23.5–41.6 kcal mol−1 above the ground state.
The light-induced triplet state 3 is generated from a MLCT transition from the HOMO of 1 having considerable 4dπ(Ru) character to LUMO having considerable π*-NO character. Such charge transfer results in the intramolecular oxidation of the Ru central atom and the concomitant change of the bonding mode of the NO ligand. In other words, the triplet state 3 must correspond to a [RuIII(NO)˙] species involving a ferromagnetic coupling of the unpaired electron of a neutral [NO]˙ (S = 1/2) ligand and the low-spin d5 RuIII metal center (S = 1/2). In effect the spin density is delocalised over the Cl(2)–Ru–N(1) framework (Scheme S2, ESI†) with Mulliken atomic spin densities of 0.89, 0.47 and 0.20 for the Ru, N(1) and Cl(2) atoms, respectively, consistent with the [RuIII(NO)˙] configuration of 3.
The [RuII(NO)+] (S = 0) → [RuIII(NO)˙] (S = 1) transformation is accompanied by the expected structural changes related with the Ru–N–O skeleton. Thus, the Ru–NO bond is significantly lengthened by 17.5 pm as a result of the dramatic decrease of the π-back donation from Ru(III) to NO ligand due to the increase of the oxidation state of ruthenium central atom and the occupation of the π*-NO MO with the transferred unpaired electron. The N–O bond is also lengthened but only by 1.7 pm. The most pronounced structural change is that related with the bending of the Ru–N–O moiety to 140.0°, which is characteristic of the [RuIII(NO)˙] configuration in nitrosylruthenium complexes. Noteworthy is the lowering of the ν(NO) stretching vibration by 141 cm−1 going from the ground to the excited triplet state. The Ru–NO bond lengthening in 3 implies labilization of the NO ligand, thus favouring the dissociative mechanism (SN1) of its substitution by the acetonitrile solvent molecule. The computed Ru–NO bond dissociation energy was predicted to be 27.6 kcal mol−1, while the exothermicity of the NO substitution reaction by the MeCN solvent molecule amounts to 8.4 kcal mol−1.
The photo-induced NO release from complex 1 proceeds easier from the two metastable states (MSI and MSII), which can be accessed by irradiation. The singlet (S = 0) MSI and MSII states lay 41.6 and 37.8 kcal mol−1 above the singlet ground state. On the other hand, the triplet (S = 1) MSI and MSII states lay 23.5 and 16.2 kcal mol−1 above the excited triplet state 3. Notice that the triplet MSI state is more stable than the singlet MSI state by 4.8 kcal mol−1, while the triplet MSII state is less stable than the singlet one by 6.3 kcal mol−1. In the singlet and triplet MSII states the NO ligand is coordinated to Ru(II) metal centre in a side-on (η2–NO) bonding mode, while in the singlet and triplet MSI states the NO ligand is coordinated in an O-end-on bonding mode. The Ru–NO bond in the singlet and triplet MSII states are lengthened by 18.7 and 47.7 pm, respectively, with respect to the corresponding bond in 1. The same holds also true for the NO bond which is strengthened by 6.5 and 7.7 kcal mol−1, respectively. The Ru–N–O skeleton is strongly bent with the Ru–N–O bond angle being only 84.3 and 72.3°, respectively. These structural features are consistent with the η2-NO bonding mode of the NO ligand in the singlet and triplet MSII states. The Ru–ON bond length in the singlet and triplet MSI states is 187.1 and 214.6 pm, respectively, whereas the N–O bond length of 118.7 and 121.0 pm closely resembles that of the free [NO]˙ ligand. It is important to notice that in the more stable triplet MSI state the Ru–O–N skeleton is bent with a Ru–N–O bond angle of 135.9°. Noteworthy is the pronounced lowering of the ν(NO) stretching vibration on going from the ground to MSII and MSI states by 439 and 104 cm−1 for the singlets, respectively, and by 534 and 225 cm−1 for the triplets, respectively. Analogous red shift of the ν(NO) stretching vibration going from the ground to MSII and MSI states has been observed experimentally in the nitroprusside ion.64 The computed Ru–NO bond dissociation energy in MSII state is 10.6 and 4.1 kcal mol−1 for the singlet and triplet, respectively, while the exothermicity of the NO substitution reaction by the MeCN solvent was predicted to be 25.6 and 31.9 kcal mol−1, respectively. Finally, the Ru–ON bond dissociation energy in the MSII state is 6.6 and 11.4 kcal mol−1 for the singlet and triplet, respectively, and the exothermicity of the NO substitution reaction by the MeCN solvent molecule was found to be 29.4 and 24.6 kcal mol−1, respectively.
3.3 Cytotoxicity properties of the cis-(Cl,Cl)-[RuCl2(NO)(terpy)]+ complex
The cells have been exposed to the compound for a relatively long time (3 days), to allow the compound to be able to reach DNA, or another biological target, and to allow it to act on this level. The complex cis-(Cl,Cl)-[RuCl2(NO)(terpy)]Cl was tested against the A2780 (human ovarian carcinoma) cell line and A2780cisR, the corresponding cisplatin-resistant cell line, that represents a good model for the screening of new anticancer metal-based agents.65The IC50 values (Table 4) for the cis-(Cl,Cl)-[RuCl2(NO)(terpy)]Cl complex (normal and resistant A2780 cell line) are 0.49 µM and 0.64 µM, respectively. These values are far better than for other Ru-pyridyl complexes, such as the antitumor-active mer-[Ru(terpy)Cl3] (IC50 = 11.0 and 32.5 µM for A2780 and A2780cisR, respectively).66 It is tempting to attribute this improvement to the presence of the nitrosyl group in cis-(Cl,Cl)-[RuCl2(NO)(terpy)]Cl complex, although a detailed molecular basis of it cannot be given and other factors, such as the solubility or structural reasons could be also responsible. Among the most active polypyridine ruthenium complexes are also compounds containing the 2-phenylazopyridine (azpy) ligand, which have been tested against the A2780 cell line.66 The complex α-[RuCl2(azpy)2] was considered as the most active, with IC50 values of 0.86 and 0.98 µM in A2780 and A2780cisR, respectively,66 which is close but not better than the present cis-(Cl,Cl)-[RuCl2(NO)(terpy)]Cl complex.
Finally, cis-(Cl,Cl)-[RuCl2(NO)(terpy)]Cl shows even a higher activity than cisplatin and carboplatin with respect to the same cell line. The tested complex is even far more active in the resistant cell line than these two platinum compounds, showing an IC50 value of 0.64 µM compared to the value of 42.8 and 2.6 µM for carboplatin and cisplatin, respectively. This further confirms that the activity of the cis-(Cl,Cl)-[RuCl2(NO)(terpy)]Cl complex is not influenced by the resistance mechanisms in the A2780cisR cell line.
3.4 DNA binding studies
![Circular dichroism spectra of CT DNA following the gradual addition of the cis-(Cl–Cl)-[RuCl2(terpy)(NO)]Cl complex. Sequence as given in the insert.](/image/article/2005/DT/b418838a/b418838a-f6.gif) | ||
| Fig. 6 Circular dichroism spectra of CT DNA following the gradual addition of the cis-(Cl–Cl)-[RuCl2(terpy)(NO)]Cl complex. Sequence as given in the insert. | ||
In the present work 31P NMR spectra of sonicated DNA, dissolved in PIPES buffer, were recorded at 37 °C, and maintaining the ionic strength, as ionic strength changes are known to have an effect on the chemical shift. The ratio between the DNA and the cis-(Cl,Cl)-[RuCl2(NO)(terpy)]Cl complex was increased from 0.1 to 0.3 and 0.5. The cis-(Cl,Cl)-[RuCl2(NO)(terpy)]Cl complex did not produce large downfield shifts in the DNA 31P signal. A small shift about 0.1 ppm was observed with a large broadening in the line width of the 31P peak as the ratio increased. At r = 0.1 ratio the compound produced a line-width broadening increased for Δν1/2 = 10 Hz and at r = 0.5 for Δν1/2 = 91 Hz in comparison with the uncomplexed DNA (Fig. 7). The broadening of the peak could be due to an increased dispersion of chemical shifts due to neighbour effects of the coordination binding of the complex to DNA. Also, the broadening may be induced by a reduction of the DNA mobility due to the binding of the complex. It is known that the binding of the cisplatin to CT-DNA gives a modest downfield shift for 31P signal, on reaction with DNA.71 On the other hand strong interactions with the phosphates of the pyridine nucleotides 5′-CMP and 5′-UMP, such as that of Et2SnCl2 in acidic media, are known to cause significant downfield 31P shifts of about 2 to 3 ppm.73,74
![23Na and 31P NMR spectra of the sonicated DNA following the addition of the cis-(Cl–Cl)-[RuCl2(terpy)(NO)]Cl complex in various ratios.](/image/article/2005/DT/b418838a/b418838a-f7.gif) | ||
| Fig. 7 23Na and 31P NMR spectra of the sonicated DNA following the addition of the cis-(Cl–Cl)-[RuCl2(terpy)(NO)]Cl complex in various ratios. | ||
To investigate the local ion exchange of Na+ and cis-(Cl,Cl)-[RuCl2(NO)(terpy)]Cl in the vicinity of B-DNA, 23Na NMR measurements have been performed under the same conditions as the 31P NMR experiments. With this spectroscopy the electrostatic interaction between the cis-(Cl,Cl)-[RuCl2(NO)(terpy)]Cl complex and the DNA was verified. A line-width narrowing from the initial DNA sample (r = 0) Δν1/2 = 23 Hz to Δν1/2 = 8 Hz at r = 0.5 was observed (Fig. 7). This effect is consistent with a perturbation of the Na+ counter ions on the DNA phosphates. The positively charged complex coordinating with the bases causes irreversible modification in the total charge of the DNA polyanion, thereby replacing the Na+ ions from the phosphates. This results in a narrowing of the sodium peak as the ratio (Ru/base) increases. Also a synergistic interaction with the DNA phosphates could not be excluded. Similar observations are known for platinum compounds, such as cis-DDP and trans-DDP.75
Agarose gel electrophoresis was applied to determine the unwinding induced in pUC9 plasmid by the complex cis-(Cl,Cl)-[RuCl2(NO)(terpy)]Cl (see Fig. 8; shows electrophoresis gels in which increasing amounts of the ruthenium complex have been added to supercoiled pUC9 DNA). Lane a contains the marker DNA from 10 000 bp—100 bp top to bottom. The plasmid pUC9 gives a single major electrophoretic band corresponding to the supercoiled form.81 Treatment with the complex cis-(Cl,Cl)-[RuCl2(NO)(terpy)]Cl results in remarkable decrease in mobility of the supercoiled form for ratios r = 0.1 to 0.5 and a slight increase when the ratio increases to r = 0.7. The supercoiled DNA at r = 0.1 (lane b) gets larger, due to unwinding of the coils in the DNA caused by the binding of cis-(Cl,Cl)-[RuCl2(NO)(terpy)]Cl and the mobility of the band in the gel is slower.
![Electrophoresis of the plasmid DNA (a) pUC9 and with the addition of the cis-(Cl–Cl)-[RuCl2(terpy)(NO)]Cl complex at ratios (b)
r
= 0.1, (c)
r
= 0.3, (d)
r
= 0.5, (e) r = 0.7, (f)
r
= 1 (r
=
[Ru]/[DNA]). Lane a contains a marker DNA from 10 000 bp–100 bp top to bottom.](/image/article/2005/DT/b418838a/b418838a-f8.gif) | ||
| Fig. 8 Electrophoresis of the plasmid DNA (a) pUC9 and with the addition of the cis-(Cl–Cl)-[RuCl2(terpy)(NO)]Cl complex at ratios (b) r = 0.1, (c) r = 0.3, (d) r = 0.5, (e) r = 0.7, (f) r = 1 (r = [Ru]/[DNA]). Lane a contains a marker DNA from 10 000 bp–100 bp top to bottom. | ||
In the next lanes (d, e) the bands are becoming more and more diffused with increasing concentration of the Ru complex. Because the plasmid DNA receives single strand the ethidium bromide cannot intercalate between the two strands anymore, so that the staining of the DNA is less and as a result the bands get fainter. At r = 1 probably the DNA of the plasmid is still covalently closed and supercoiled, but the nucleotides are not paired anymore. No bands are visible and there is no evidence that the DNA is relaxed or linear.
Since the complex cis-(Cl,Cl)-[RuCl2(NO)(terpy)]Cl has at least two active coordination sites, containing two labile Cl ions, it could be possible that it forms DNA intrastrand cross-links. However, it has been reported that octahedral Ru(III) complexes appear to be sterically unfavourable for such type of bifunctional binding.82 Thus, the band that corresponds to DNA–Ru complex moves relatively faster than the band for DNA alone, which indicates that the ruthenium binding induces uncoiling of the superhelix of the plasmid DNA.
The interaction of antitumor-active ruthenium complexes, such as Na[trans-RuCl4(dmso)(Him)] (NAMI) and dichloro(1,2-propylenediaminetetraacetate)ruthenium(III) (RAP) with supercoiled DNA shows an alteration in the DNA conformation and a modification in the electrophoretic mobility.68,83 Also, cisplatin causes a large decrease in the mobility of the supercoiled form as the amount of the added complex increases.80
![Electrophoresis of the plasmid DNA digested with the enzymes DraI and SmaI and without restriction enzymes (a) DNA at ratios (b)
r
= 0.1, (c)
r
= 0.3, (d) fresh DNA (r
=
[Ru]/[base pair]). The asterisks in the figure refer to the weak band not recognized by the restriction enzyme.](/image/article/2005/DT/b418838a/b418838a-f9.gif) | ||
| Fig. 9 Electrophoresis of the plasmid DNA digested with the enzymes DraI and SmaI and without restriction enzymes (a) DNA at ratios (b) r = 0.1, (c) r = 0.3, (d) fresh DNA (r = [Ru]/[base pair]). The asterisks in the figure refer to the weak band not recognized by the restriction enzyme. | ||
4 Concluding remarks
The spectroscopic and analytical data presented above clearly indicate that the complex resulting from the reaction between the [RuCl3(H2O)2(NO)] and terpyridine is the cis-(Cl,Cl)-[RuCl2(terpy)(NO)]Cl. The release of nitric oxide upon irradiation with a mercury lamp in acetonitrile solution is demonstrated and analysed theoretically. Intermediate metastable states MSI and MSII either in their singlet or triplet states, would finally yield the NO free cis-(Cl,Cl)-[RuCl2(terpy)(MeCN)] species. DFT calculations have provided a satisfactory description of structural, bonding, electronic and related properties (such as the kinetics84) of the new compound and provide insight in to the mechanism of the photo-induced NO release.The predicted relatively weak association of the solvent molecule permits its easy dissociation into [RuIIICl2(terpy)]+ cationic species which might be the bioactive species capable to coordinate with the guanine and cytosine DNA bases.
Screening on A2780 (human ovarian carcinoma) cell lines has shown a good cytotoxicity with IC50 values as low as 0.49 µM, i.e. much lower than [RuCl3(terpy)] and cisplatin or carboplatin. 31P and 23Na NMR spectroscopy together with electrophoretic mobility studies indicate that the complex coordinates to DNA, assisted by electrostatic interactions. Studies on the inhibition of the restriction enzymes DraI and SmaI have shown that the complex prefers the guanine and cytosine DNA bases.
Acknowledgements
This work was supported by a Marie Curie Training Fellowship from the EU in the 5th Framework programme (MEDICINOR; Grant No. HPMT-CT-2000-00192), allowing one author (KK) to spend time at the LIC in Leiden to perform most of the experimental work, Also the Greek General Secretariat of Research and Technology is thanked for financial support (PENED 2001).References
- E. Culotta and D. E. Koshland, Jr., Science, 1992, 258, 1862.
- A. Calver, J. Collier and P. Vallance, Exp. Physiol., 1993, 78, 303 Search PubMed.
- L. J. Ignarro, Hypertension, 1990, 16, 477 Search PubMed.
- S. H. Snyder and D. S. Bredt, Sci. Am., 1992, 266, 68 CrossRef CAS.
- K. Shibuki and D. Okada, Nature, 1991, 349, 326 CrossRef CAS.
- G. Stochel, A. Wanat, E. Kulis and Z. Stasicka, Coord. Chem. Rev., 1998, 171, 203 CrossRef CAS.
- E. Tfouni, M. Krieger, B. R. McGarvey and D. W. Franco, Coord. Chem. Rev., 2003, 236, 57 CrossRef CAS.
- D. R. Lang, J. A. Davis, L. G. F. Lopes, A. A. Ferro, L. C. G. Vasconcellos, D. W. Franco, E. Tfouni, A. Wieraszko and M. J. Clarke, Inorg. Chem., 2000, 39, 2294 CrossRef CAS.
- N. Bettache, T. Carter, J. E. T. Corrie, D. Ogden and D. R. Trentham, Methods Enzymol., 1996, 268, 266 CAS.
- T. D. Carter, N. Bettache and D. Ogden, Br. J. Pharmacol., 1997, 122, 971 CrossRef CAS.
- J. M. Slocik and R. E. Shepherd, Inorg. Chim. Acta, 2000, 311, 80 CrossRef CAS.
- P. C. Ford, J. Bourassa, K. Miranda, B. Lee, I. Lorkovic, S. Boggs, S. Kudo and L. Laverman, Coord. Chem. Rev., 1998, 171, 185 CrossRef CAS.
- R. Ackroyd, C. Kelty, N. Brown and M. Reed, Photochem. Photobiol., 2001, 74, 656 CAS.
- M. G. Sauaia, F. D. Oliveira, A. C. Tedesco and R. S. da Silva, Inorg. Chim. Acta, 2003, 355, 191 CrossRef CAS.
- A. Y. Ershov, A. D. Shashko, O. V. Sizova and N. V. Ivanova, Russ. J. Gen. Chem., 2002, 72, 1327 CrossRef CAS.
- V. Togniolo, R. S. da Silva and A. C. Tedesco, Inorg. Chim. Acta, 2001, 316, 7 CrossRef CAS.
- O. V. Sizova, V. V. Sizov, A. Y. Ershov, A. B. Nikol'skii, V. L. Baranovskii and A. D. Shashko, Russ. J. Gen. Chem., 2001, 71, 995 CrossRef CAS.
- H. Nagao, D. Ooyama, T. Hirano, H. Naoi, M. Shimada, S. Sasaki, N. Nagao, M. Mukaida and T. Oi, Inorg. Chim. Acta, 2001, 320, 60 CrossRef CAS.
- T. Hirano, K. Ueda, M. Mukaida, H. Nagao and T. Oi, J. Chem. Soc., Dalton Trans., 2001, 2341 RSC.
- H. Nagao, N. Nagao, Y. Yukawa, D. Ooyama, Y. Sato, T. Oosawa, H. Kuroda, F. S. Howell and M. Mukaida, Bull. Chem. Soc. Jpn., 1999, 72, 1273 CrossRef CAS.
- M. Mukaida, Y. Sato, H. Kato, M. Mori, D. Ooyama, H. Nagao and F. S. Howell, Bull. Chem. Soc. Jpn., 2000, 73, 85 CrossRef CAS.
- A. Dovletoglou, S. A. Adeyemi and T. J. Meyer, Inorg. Chem., 1996, 35, 4120 CrossRef.
- D. W. Pipes and T. J. Meyer, Inorg. Chem., 1984, 23, 2466 CrossRef CAS.
- R. W. Callahan and T. J. Meyer, Inorg. Chem., 1977, 16, 574 CrossRef CAS.
- S. A. Adeyemi, T. J. Meyer and F. J. Miller, Inorg. Chem., 1972, 11, 994 CrossRef CAS.
- O. Novakova, J. Kasparkova, O. Vrana, P. M. Vanvliet, J. Reedijk and V. Brabec, Biochemistry, 1995, 34, 12369 CrossRef CAS.
- P. M. Van Vliet, J. G. Haasnoot and J. Reedijk, Inorg. Chem., 1994, 33, 1934 CrossRef CAS.
- F. Zobi, M. Hohl, I. Zimmermann and R. Alberto, Inorg. Chem., 2004, 43, 2771 CrossRef CAS.
- J. Reedijk, Curr. Opin. Chem. Biol., 1999, 3, 236 CrossRef CAS.
- W. D. Wilson, F. A. Tanious, M. Fernandez-Saiz, C. T. Rigl and K. R. Fox, Methods in Molecular Biology, 1997, vol. 90, Humana Press, Clifton, NJ, USA Search PubMed.
- J. M. Fletcher, I. L. Jenkins, F. M. Lever, F. S. Martin, A. R. Powell and R. Todd, J. Inorg. Nucl. Chem., 1955, 1, 378 CrossRef CAS.
- A. Catte, F. C. Marincola, M. Casu, G. Saba and A. Lai, J. Biomol. Struct. Dyn., 2002, 20, 99 Search PubMed.
- H. Tada, O. Shiho, K. Kuroshima, M. Koyama and K. Tsukamoto, J. Immunol. Methods, 1986, 93, 157 CrossRef CAS.
- B. A. J. Jansen, P. Wielaard, H. den Dulk, J. Brouwer and J. Reedijk, Eur. J. Inorg. Chem., 2002, 2375 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scusseria, M. A. Robb, J. R. Cheeseman, J. A. Montgomery, T. Vreven, K. N. Kudin, J. C. Burant, J. M. Millam, S. S. Iyengar, J. Tomasi, V. Barone, B. Mennucci, M. Cossi, G. Scalmani, N. Rega, G. A. Petersson, H. Nakatsuji, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, M. Klene, X. Li, J. E. Knox, H. P. Hratchian, J. B. Cross, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, P. Y. Ayala, K. Morokuma, G. A. Voth, P. Salvador, J. J. Dannenberg, V. G. Zakrzewski, S. Dapprich, A. D. Daniels, M. C. Strain, O. Farkas, D. K. Malick, A. D. Rabuck, K. Raghavachari, J. B. Foresman, J. V. Ortiz, Q. Cui, A. G. Baboul, S. Clifford, J. Cioslowski, B. B. Stefanov, G. Liu, A. Liashenko, P. Piskorz, I. Komaromi, R. L. Martin, D. J. Fox, T. Keith, M. A. Al-Laham, C. Y. Peng, A. Nanayakkara, M. Challacombe, P. M. W. Gill, B. Johnson, W. Chen, M. W. Wong, C. Gonzalez, and J. A. Pople, Gaussian 03, Revision B.02, Gaussian, Inc.Pittsburgh P.A., 2003 Search PubMed.
- A. D. Becke, J. Chem. Phys., 1992, 96, 2155 CrossRef CAS.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648 CrossRef CAS.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B, 1998, 37, 785 Search PubMed.
- D. Andrae, U. Haussermann, M. Dolg, H. Stoll and H. Preuss, Theor. Chim. Acta, 1990, 77, 123 CAS.
- N. Godbout, D. R. Salahub, J. Andzelm and E. Wimmer, Can. J. Chem.-Rev. Can. Chim., 1992, 70, 560 Search PubMed.
- W. R. Wadt and P. J. Hay, J. Chem. Phys., 1985, 82, 284 CrossRef CAS.
- P. J. Hay and W. R. Wadt, J. Chem. Phys., 1985, 82, 299 CrossRef CAS.
- H. B. Schlegel, J. Comput. Chem., 1982, 3, 214 CrossRef CAS.
- F. Weinhold, The Encyclopedia of Computational Chemistry; ed. P. v. R. Schleyer, John Wiley & Sons, Chichester, 1998, 1792 Search PubMed.
- S. J. A. van Gisbergen, F. Kootstra, P. R. T. Schipper, O. V. Gritsenko, J. G. Snijders and E. J. Baerends, Phys. Rev. A, 1998, 57, 2556 CrossRef CAS.
- R. Bauernschmitt and R. Ahlrichs, Chem. Phys. Lett., 1996, 256, 454 CrossRef CAS.
- C. Jamorski, M. E. Casida and D. R. Salahub, J. Chem. Phys., 1996, 104, 5134 CrossRef CAS.
- P. Boulet, H. Chermette, C. Daul, F. Gilardoni, F. Rogemond, J. Weber and G. Zuber, J. Phys. Chem. A, 2001, 105, 885 CrossRef CAS.
- A. Rosa, E. J. Baerends, S. J. A. van Gisbergen, E. van Lenthe, J. A. Groeneveld and J. G. Snijders, J. Am. Chem. Soc., 1999, 121, 10356 CrossRef CAS.
- N. N. Nevskii, N. M. Sinitsyn and A. A. Svetlov, Zh. Neorg. Khim., 1990, 35, 1159 Search PubMed.
- J. R. During, W. A. McAllister, J. N. Willis and E. E. Mercer, Spectrochim. Acta, 1966, 22, 1091 CrossRef.
- J. B. Godwin and T. J. Meyer, Inorg. Chem., 1971, 10, 471 CrossRef.
- H. Nagao, H. Nishimura, H. Funato, Y. Ichikawa, F. S. Howell, M. Mukaida and H. Kakihana, Inorg. Chem., 1989, 28, 3955 CrossRef CAS.
- G. M. Bryant, Fergusson Je and H. K. J. Powell, Aust. J. Chem., 1971, 24, 257 CAS.
- A. K. Patra and P. K. Mascharak, Inorg. Chem., 2003, 42, 7363 CrossRef CAS.
- J. Bordini, D. L. Hughes, J. D. D. Neto and C. J. da Cunha, Inorg. Chem., 2002, 41, 5410 CrossRef CAS.
- J. A. Olabe, L. A. Gentil, G. Rigotti and A. Navaza, Inorg. Chem., 1984, 23, 4297 CrossRef CAS.
- J. Enemark and R. Feltham, Coord. Chem. Rev., 1974, 13, 339 CrossRef CAS.
- G. B. Richter-Addo and P. Legzdins, Metal Nitrosyls, Oxford University Press, New York, 1992 Search PubMed.
- K. Nakamoto, Infrared and Raman Spectra of Inorganic and Coordination Compounds, 3rd edn., Wiley, New York, 1978, p. 222 Search PubMed.
- P. Coppens, I. Novozhilova and A. Kovalevsky, Chem. Rev., 2002, 102, 861 CrossRef CAS.
- S. C. Da Silva and D. W. Franco, Spectrochim. Acta, Part A., 1999, 55, 1515 CrossRef.
- M. D. Carducci, M. R. Pressprich and P. Coppens, J. Am. Chem. Soc., 1997, 119, 2669 CrossRef CAS.
- J. A. Guida, P. J. Aymonino, O. E. Piro and E. E. Castellano, Spectrochim. Acta, Part A., 1993, 49, 535 CrossRef.
- J. Reedijk, J. Inorg. Biochem., 2003, 96, 81 CrossRef.
- A. C. G. Hotze, M. Bacac, A. H. Velders, B. A. J. Jansen, H. Kooijman, A. L. Spek, J. G. Haasnoot and J. Reedijk, J. Med. Chem., 2003, 46, 1743 CrossRef CAS.
- V. Brabec, V. Kleinwachter, J. L. Butour and N. P. Johnson, Biophys. Chem., 1990, 35, 129 CrossRef CAS.
- E. Gallori, C. Vettori, E. Alessio, F. G. Vilchez, R. Vilaplana, P. Orioli, A. Casini and L. Messori, Arch. Biochem. Biophys., 2000, 376, 156 CrossRef CAS.
- T. R. Krugh and M. E. Nuss, Biological Applications of Magnetic Resonance, ed. R. G. Shulman, Academic Press, New York, 1979, 113 Search PubMed.
- D. G. Gorenstein, Annu. Rev. Biophys. Bioeng., 1981, 10, 355 Search PubMed.
- W. D. Wilson, B. L. Heyl, R. Reddy and L. G. Marzilli, Inorg. Chem., 1982, 21, 2527 CrossRef CAS.
- W. D. Wilson and R. L. Jones, Adv. Pharmacol. Chemother., 1981, 18, 177 Search PubMed.
- L. Ghys, M. Biesemans, M. Gielen, A. Garoufis, N. Hadjiliadis, R. Willem and J. C. Martins, Eur. J. Inorg. Chem., 2000, 513 CrossRef CAS.
- Z. Yang, T. Bakas, A. Sanchez-Diaz, C. Charalampopoulos, J. Tsangaris and N. Hadjiliadis, J. Inorg. Biochem., 1998, 72, 133 CrossRef CAS.
- G. Mallet, S. Ansiss and D. Vasilescu, J. Biomol. Struct. Dyn., 1998, 16, 21 Search PubMed.
- G. A. Neyhart, N. Grover, S. R. Smith, W. A. Kalsbeck, T. A. Fairley, M. Cory and H. H. Thorp, J. Am. Chem. Soc., 1993, 115, 4423 CrossRef CAS.
- E. Tselepikalouli and N. Katsaros, J. Inorg. Biochem., 1989, 37, 271 CrossRef CAS.
- R. B. Nair, E. S. Teng, S. L. Kirkland and C. J. Murphy, Inorg. Chem., 1998, 37, 139 CrossRef CAS.
- G. L. Cohen, W. R. Bauer, J. K. Barton and S. J. Lippard, Science, 1979, 203, 1014 CAS.
- S. E. Sherman and S. J. Lippard, Chem. Rev., 1987, 87, 1153 CrossRef CAS.
- M. Mottes, G. Grandi, V. Sgaramella, U. Canosi, G. Morelli and T. A. Trautner, Mol. Gen. Genet., 1979, 174, 281 CrossRef CAS.
- G. Esposito, S. Cauci, F. Fogolari, E. Alessio, M. Scocchi, F. Quadrifoglio and P. Viglino, Biochemistry, 1992, 31, 7094 CrossRef CAS.
- M. Bacac, A. C. G. Hotze, K. van der Schilden, J. G. Haasnoot, S. Pacor, E. Alessio, G. Sava and J. Reedijk, J. Inorg. Biochem., 2004, 98, 402 CrossRef CAS.
- J. Reedijk, Proc. Natl. Acad. Sci. USA, 2003, 100, 3611 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Table S1: Selected bond lengths (pm), bond angles (°), and unscaled ν(N–O) harmonic vibrational frequencies (cm−1) of the cis- and trans-(Cl,Cl)-[Ru(terpy)(NO)Cl2]+ complexes calculated at the B3LYP level and comparison with available experimental data. Table S2: GIAO-B3LYP/SDD 13C, 15N, 17O and 1H NMR absolute isotropic shielding tensor elements (σ, ppm) of the cis- and trans-(Cl,Cl)-[Ru(terpy)(NO)Cl2]+ complexes Computed at the B3LYP/SDD level. Table S3: GIAO-B3LYP/SDD 13C, 15N, 17O and 1H NMR absolute isotropic shielding tensor elements (σ, ppm) of TMS, nitromethane and formamide compounds computed at the B3LYP/SDD level. Scheme S2: The 13C, 15N and 1H chemical shifts for cis- and trans-dichloro(nitrosyl)(terpyridine)ruthenium(II) cation. See http://www.rsc.org/suppdata/dt/b4/b418838a/ |
| This journal is © The Royal Society of Chemistry 2005 |