Substituent effects of iridium complexes for highly efficient red OLEDs
Shinjiro
Okada
,
Keiji
Okinaka
,
Hironobu
Iwawaki
,
Manabu
Furugori
,
Masashi
Hashimoto
,
Taihei
Mukaide
,
Jun
Kamatani
,
Satoshi
Igawa
,
Akira
Tsuboyama
,
Takao
Takiguchi
and
Kazunori
Ueno
*
OD project Canon Inc., 5-1, Morinosato Wakamiya, Atsugishi, Kanagawa, Japan. E-mail: ueno.kazunori@canon.co.jp; okada.shinjiro@canon.co.jp
First published on 24th March 2005
Abstract
This study reports substituent effects of iridium complexes with 1-phenylisoquinoline ligands. The emission spectra and phosphorescence quantum yields of the complexes differ from that of tris(1-phenylisoquinolinato-C2,N)iridium(III) (Irpiq) depending on the substituents. The maximum emission peak, quantum yield and lifetime of those complexes ranged from 598–635 nm, 0.17–0.32 and 1.07–2.34 µs, respectively. This indicates the nature of the substituents has a significant influence on the kinetics of the excited-state decay. The substituents attached to phenyl ring have an influence on a stability of the HOMO. Furthermore, those substituents have effect on the contribution to a mixing between 3π–π* and 3MLCT for the lowest excited states. Some of the complexes display the larger quantum yield than Irpiq, which has the quantum yield of 0.22. The organic light emitting diode (OLED) device based on tris [1-(4-fluoro-5-methylphenyl)isoquinolinato-C2,N]iridium(III) (Ir4F5Mpiq) yielded high external quantum efficiency of 15.5% and a power efficiency of 12.4 lm W−1 at a luminance of 218 cd m−2. An emission color of the device was close to an NTSC specification with CIE chromaticity characteristics of (0.66, 0.34).
Introduction
The development of the first thin film hetero-junction organic light emitting diode in 1987 by Tang and VanSlyke1 had stimulated the research in OLEDs for practical applications using fluorescent materials, for the last 15 years. Especially, a lot of efforts have been concentrated on developing new materials with high quantum yields, for the realization of higher efficiency. Recently, OLEDs using phosphorescent materials are attracting great interest due to its potential in the application of flat panel display. Iridium(III) complexes with 2-phenylpyridine are well known to exhibit high triplet quantum yields due to the mixing of the singlet excited state and the triplet excited state via spin–orbit coupling, which leads to high phosphorescence yields, enhancing the triplet state.2–4 The goal of material research is to achieve high power efficiency and low power consumption when OLED display is used in mobile appliance.5–8Our previous reports on electroluminescence from Ir complex with 1-phenylisoquinoline as the ligands, (Irpiq: Tris(1-phenylisoquinolinato-C2,N)iridium(III)) doped into a 4,4′-bis(carbazole-9-yl)biphenyl (CBP) emphasized its dramatically increased power efficiency of 6.8 lm W−1 (8.2 cd A−1) at 100 cd m−2 and excellent color CIE characteristics (0.68, 0.32) for red.9,10
The substitution effect on 2-phenylpyridine ligand of iridium,11–13 cyclometalating ligands of platinum complexes14 and iridium complexes,15,16 diimine ligands of platinum complexes17 and the effect of phenanthroline ligand substituents on radiative and nonradiative process concerning ruthenium(II) and iridium(III) complexes had been also reported previously.18Y. J. Su et al. had reported the study on fluorine substituted Irpiq.19 In the present work we report the effect of a series of substituent on the phenyl ring of Irpiq. Also the luminescence, quantum yield, lifetime, analysis of luminescing state and the performance of organic EL device using these complexes are discussed in detail. These results enable us to expand our knowledge of fine tuning capabilities of ligand substituents and establish a guideline for designing novel red phosphorescent molecules.
Experimental
NMR spectra were recorded on a Bruker model DPX-400 (1H: 400 MHz, 13C: 100 MHz) or a Bruker model AVANCE-500 (13C: 125 MHz) NMR spectrometer. Mass spectra data were measured by JEOL JMS-700 operating in electron impact (EI). Elemental analysis was carried out with an elemental analyzer Vario EL CHNOS from Elementar Co.Crystals of iridium complex suitable for X-ray analysis were obtained from chloroform solutions at room temperature. Diffraction data were collected at 93.1 on a Rigaku RAXIS-RAPID imaging plate diffractometer equipped with graphite-monochromated Mo Kα radiation (λ = 0.71069 Å). The crystal structure was solved by the direct method using SIR92.20 The crystal structure was refined by the full-matrix least-squares method on F2. All non-hydrogen atoms were refined anisotropically, while hydrogen atoms were refined isotropically. All analyses were performed by the crystallographic software package CrystalStructure 3.6.0.21
[C45H27N3F3Ir][CHCl3], M 978.32 g mol−1, T 93.1 K, monoclinic, P21/n, Z 4, a 13.3262(4), b 19.4561(4), c 14.3648(3) Å, β 96.785(1)°, V 3698.4(2) Å3, µ(MoKα)/mm−1, graphite monochromator, 10795 reflections used, R 0.032, Rw 0.006.
CCDC reference number 240125.
See http://www.rsc.org/suppdata/dt/b4/b417058j/ for crystallographic data in CIF or other electronic format.
Photoluminescence spectra were recorded on a Hitachi F4500 fluorescence spectrometer using 464 nm excitation pulse. Spectral data were corrected with the use of emission spectra of tris(2-phenylpyridinato-C2,N)iridium(III) (fac-Irppy) and fac-Irpiq measured by Spectroradiometer SR1 from TOPCON Ltd. Phosphorescence quantum yields were measured at room temperature using an N2-saturated toluene solution of 10−5 M. The quantum yields were determined using fac-Irppy as a reference (ϕ = 0.4).22
Emission lifetime measurements were carried out using a Hamamatsu Photonics C4334 streakscope with excitation light (λ = 420 nm) from a N2 laser (LN120C from Laser Photonics). UV-visible spectra were recorded on a Shimadzu UV3100S spectrophotometer.
The cyclic voltammograms of the complexes were measured in CH2Cl2 (Eox½), THF (Ered½) solutions containing n-Bu4NClO4 (0.1 M) at a 20 mV s−1 scan rate using a Pt rod (Eox½) working electrode, a glassy carbon (Ered½) working electrode, a Pt rod counter electrode, and an Ag/Ag+ reference electrode under an N2 atmosphere. All potentials are reported against an Ag/Ag+ reference.
Synthesis, general procedure
All ligands were prepared through Suzuki coupling. Ligands with isoquinoline core were synthesized by the reaction of corresponding halogenated isoquinoline and aryl boronic acids. Other ligands with substituted 1-phenylisoquinolines were synthesized by analogous reactions. The ligand compounds were purified by silica gel column chromatography with CHCl3 as an eluent. Phenyl isoquinoline ligands were prepared from isoquinoline N-oxide as a precursor. The synthesis scheme of 1-phenylisoquinoline is given below (Scheme 1).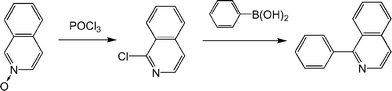 | ||
| Scheme 1 | ||
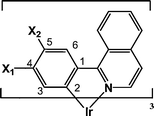
Complexes were synthesized according to a previous paper.13 Ir(acac)3 [iridium(III) acetylacetonate] (0.5 g, 1.0 mmol) and cyclometalating ligand (ca. 5 mmol) were dissolved in 50 ml glycerol. The solution was refluxed under nitrogen stream for 6 h. After completion of the reaction, addition of 1 M HCl resulted in precipitation of the product, which was filtered, washed with water, and dried at 100 °C in vacuum. The product was purified by silica gel column chromatography with CHCl3 as an eluent. Yields of the products were in the range from 11 to 45% based on Ir(acac)3. Molecular structures of the complexes in this paper and their abbreviations are listed in Table 1.
|
|
|||
|---|---|---|---|
| No. | Complex | Attached position | |
| X1 | X2 | ||
| 1 | Irpiq | ||
| 2 | Ir4Fpiq | F | |
| 3 | Ir4F5Mpiq | F | Methyl |
| 4 | Ir4MOpiq | Methoxy | |
| 5 | IrC4piq | Butyl | |
| 6 | Ir4Mpiq | Methyl | |
| 7 | Ir5iPrpiq | Isopropyl | |
| 8 | Ir5Mpiq | Methyl | |
The 1H NMR spectra and elemental analysis data of the complexes 1 to 8 are as follows.
OLED fabrication and evaluation
The device geometry of the OLED is shown in Fig. 1. | ||
| Fig. 1 Device structure and the molecular structures of the compounds used in the device. | ||
The organic materials were deposited by thermal evaporation at pressures of less than 10−4 Pa. The devices were fabricated on indium tin oxide (ITO) film (150 kΩ m−2, thickness 120 nm, from Nippon Sheet Glass Co.) with a 3.14 mm2 round-patterned area. A four layer device employed an ITO as an anode, a 40 nm thick α-NPD (4,4′-bis(N-1-naphthyl-N-phenylamino)biphenyl) as the hole transport material, Ir4F5Mpiq (3) as the luminescent material, CBP (4,4′-bis(carbazole-9-yl)biphenyl) as a host material of a 20 nm thick light emitting layer, a 50 nm thick Bphen (4,7-diphenyl-1,10-phenanthroline) as the electron transport material, a 1 nm thick potassium fluoride as an electron injection layer and 150 nm thick Al as the cathode. The device structure and the structural materials are shown in Fig. 1. The light-emitting layer was prepared by coevaporating a host (CBP) and a phosphorescent material as a dopant, with both deposition rates being controlled with two independent quartz crystal oscillators. The doping concentration of the dopant was a 10 wt%.
After the device fabrication, they were encapsulated with calcium oxide as desiccant. Current–voltage characteristics of the devices were measured by an automated homemade set up consisting a waveform generator Yokogawa AG1200, and a digital oscilloscope TEKTRONIKS TDS420. A luminance colorimeter BM-7 from TOPCON Ltd was used for measuring the luminance of the OLED.
Results and discussion
(1) Characterization of the complexes
The iridium center of Ir4Fpiq (2) is octahedrally coordinated by three bidentate ligands with a facial structure and a 3-fold rotational axis passing through the iridium atom.A molecular plot of Ir4Fpiq (2) is shown in Fig. 2. Some selected bond lengths (Å) and angles (°) for Ir4Fpiq (2) and Irpiq (1) are presented in Table 2. The detailed characteristic data of Irpiq (1) had been reported.10 All bonds and angles of those two complexes are very similar. The two complexes have facial octahedral structure. The 1H NMR spectra of all complexes 1 to 8 are consistent with the facial structure, which indicates that the number of coupled spins is equal to that of protons on one ligand because the three ligands are magnetically equivalent due to the 3-fold symmetry.
| Irpiq (1) | Ir4Fpiq (2) | ||
|---|---|---|---|
| Bond lengths/Å | Ir(1)–N(1) | 2.135(5) | 2.134(3) |
| Ir(1)–C(15) | 2.009(6) | 2.006(3) | |
| N(1)–C(1) | 1.374(8) | 1.364(4) | |
| N(1)–C(9) | 1.339(8) | 1.348(4) | |
| Bond angles/° | N(1)–Ir(1)–C(15) | 78.5(2) | 78.4(1) |
| Ir(1)–N(1)–C(1) | 124.8(4) | 123.5(2) | |
| Ir(1)–N(1)–C(9) | 115.1(4) | 116.1(2) | |
| Ir(1)–C(15)–C(10) | 116.3(4) | 115.8(2) | |
| C(1)–N(1)–C(9) | 119.7(5) | 120.3(3) | |
 | ||
| Fig. 2 ORTEP drawing of Ir4Fpiq (2). The thermal ellipsoids representing a 50% probability level. Hydrogen atoms are omitted for clarity. | ||
The absorption and photoluminescence (PL) properties of the newly synthesized iridium complexes are summarized in Table 3. The complexes have quantum yield ranging from 0.17 to 0.32, emission maximum peak (λmax)), ranging from 598 to 635 nm and lifetimes ranging from 1.07 to 2.34 µs, indicating that the nature of the substituents and therefore ligands have a significant influence on the kinetics of the excited-state decay. A maxmum peak of the phosphorescent spectrum and phosphorescent life time of Ir4Fpiq (2) were consistent with the observation of Y. J. Su et al.19
| No. | Complex | Quantum yield Φa | Lifetime τ/µs | k r b × 103/s−1 | k nr c × 103/s−1 | Absorptiongλ/nm (εmold × 10−3/M−1 cm−1) | λ max e , g/nm | Hammett constantf |
|---|---|---|---|---|---|---|---|---|
| a Quantum yield at room temperature in toluene below 10−6 M measured with 464 nm excitation Relative value on the basis of Irppy as the quantum yield is 0.4. b Radiative rate constant calculated from kr = Φ/τ. c Nonradiative rate constant calculated from knr = 1/ τ − kr. d Molar absorbance coefficient. e Corrected maximum emission peak by PMA11 (Hamamatsu Photonics). f Hammett constant (σp, σm).24,25,26 g Absorption and emission peak, λmax, were measured in toluene solution. s Shoulder peak. | ||||||||
| 1 | Irpiq | 0.22 | 1.12 | 194.2 | 698.2 | 430(15), 477(11), 544(4), 595(2) | 621 | 0.00 |
| 2 | Ir4Fpiq | 0.32 | 2.34 | 138.2 | 288.6 | 404(19), 456(12), 568(1) | 598 | 0.34 |
| 3 | Ir4F5Mpiq | 0.26 | 1.65 | 154.7 | 451.4 | 419(17), 471(10), 518(2)s | 607 | 0.17 |
| 4 | Ir4MOpiq | 0.23 | 1.81 | 125.0 | 426.3 | 418(29), 469(15), 516(4)s | 610 | 0.12 |
| 5 | IrC4piq | 0.25 | 1.36 | 187.2 | 550.8 | 428(17), 476(11), 550(4), 598(1) | 620 | −0.08 |
| 6 | Ir4Mpiq | 0.26 | 1.29 | 203.2 | 569.6 | 428(18), 475(13), 534(5), 589(2) | 621 | −0.07 |
| 7 | Ir5iPrpiq | 0.17 | 1.07 | 161.4 | 777.6 | 446(16), 456(13), 534(5), 589(2) | 635 | −0.15 |
| 8 | Ir5Mpiq | 0.17 | 0.97 | 177.7 | 853.20 | 444(15), 486(12), 558(4), 606(2) | 635 | −0.17 |
Hartree–Fock calculations were carried out on Irpiq (1), Ir4Fpiq (2) and Ir4F5Mpiq (3) using the Gaussian98 software package (Gaussian, Inc.)23 with a LanL2DZ basis set.14,24–27 The Hartree–Fock calculations suggest that the highest occupied molecular orbital (HOMO) of the three complexes are mainly localized on the phenyl ring and iridium center. On the contrary, the lowest unoccupied molecular orbital (LUMO) is mainly localized on the isoquinoline ring portion of the cyclometalating ligand (Fig. 3). The orbital picture predicts that variation in the electronic properties of the ligands should have an effect on the energy of the excited state. The calculated HOMOs of Irpiq (1), Ir4Fpiq (2) and Ir4F5Mpiq (3) were −7.37, −7.63 and −7.83 eV, respectively. And the LUMOs of the above mentioned complexes were 1.57, 1.27 and 1.34 eV, respectively (shown in Fig 3, inset).
 | ||
| Fig. 3 Hartree–Fock results of HOMO (left) and LUMO (right) for Irpiq (1). The HOMO is mainly localized on the phenyl ring and iridium and the LUMO is localized on the isoqunoline ring mainly. | ||
(2) Substituent effect of phenyl ring on HOMO
Table 4 shows electrochemical data of the complexes 1–6 which have substituents on phenyl ring. It is obvious that the HOMO level of the compounds 1–6 is strongly affected by a kind of substituent on the phenyl ring. Especially, the oxidation potential of the electron-withdrawing group ‘fluorine’ [4Fpiq (2), 4F5Mpiq (3)] is higher in comparison with the electron donating alkyl groups [C4piq (5), 4Mpiq (6)]. On the other hand, substituent effect on the LUMO is very small. As given in Table 4, the difference of the reduction potential between the highest 4Fpiq (2) and the lower 4Mpiq (6) was only 0.17 V. The oxidation and reduction potentials increase along the series Irpiq (1) < Ir4F5Mpiq (3) < Ir4Fpiq (2).| No. | Complex | Hammett constant | E ox ½/V (ΔE/mV)a | E red ½/V (ΔE/mV)a | ||
|---|---|---|---|---|---|---|
| a E ox ½ and Ered½ are the oxidation and reduction potentials between peaks, respectively. ΔE is the peak potential difference. | ||||||
| 1 | Irpiq | 0 | 0.45 (60) | −2.18 (70) | ||
| 2 | Ir4Fpiq | 0.34 | 0.65 (70) | −2.08 (90) | ||
| 3 | Ir4F5Mpiq | 0.17 | 0.54 (60) | −2.17 (110) | ||
| 4 | Ir4MOpiq | 0.12 | 0.43 (70) | −2.28 (100) | ||
| 5 | IrC4piq | −0.08 | 0.36 (60) | −2.18 (90) | ||
| 6 | Ir4Mpiq | −0.07 | 0.44 (120) | −2.25 (120) | ||
| 7 | Ir5iPrpiq | −0.15 | 0.36 (310) | N.d. (—) | ||
| 8 | Ir5Mpiq | −0.17 | 0.32 (80) | N.d. (—) | ||
These results are in agreement with the calculation (Fig. 3). As seen from Table 4, electron donating substituent on phenyl ring such as alkyl group is effective to raise the energy of the HOMO (the Eox½ is lower) and consequently reducing the energy gap, while the phenyl rings with electron-withdrawing substituents such as fluorine lowers the energy of the HOMO to make the energy gap wider. The emission energies increase along the series, 4-fluoro(4F) > 4-fluoro-5-methyl (4F5M) > 4-methoxy (4MO) > 4-butyl (C4) > H > 4-methyl (4M) > 5-isopropyl (5iPr) > 5-methyl (5M) as shown in Table 3. This tendency arises from the substituent effect of the d orbital (t2g) stabilization of iridium metal through the carbon atom–iridium bonding. And this tendency identifies with the inductive trend of the substituents. Therefore the HOMO stability and the emission energy gap are controlled by the nature of substituents and its inductive influence on the aromatic ring.
(3) A relationship between HOMO and Hammett constant
Skarda et al. had explained the substitution effect of 4,4′- and 5,5′-disubstituted 2,2′-bipyridyls by plotting the reduction potential, Ered½, against the Hammett constant σ.28 We tried to quantify the substitution effect for HOMO level by plotting oxidation potential Eox½ against the Hammett constant σ (Fig. 4). In the case of double substitution at meta- and para-positions on a phenyl ring, the sum of both σm and σp was applied.28–31 It is clear from Fig. 4 that there exists a good correlation between oxidation potentials of iridium complexes and the σs of substituents on their ligands were obtained. In this work we proposed the HOMO level of the complexes could be rationalized to Hammett constant. The emission energy from a series of the Ir complexes, 1–8versus Hammett constants is plotted in Fig. 5, which shows a linear relation between the emission energy and Hammett constant (Line A). The ligands that have the substituent with the larger Hammett constant (electron withdrawing substituents) lead to large emission energy by the lowering of the HOMO. On the contrary, the smaller Hammett constant (electron donating substituents) leads to a smaller emission energy by raising the HOMO, as shown in Fig. 5(b). Thus, by using Fig. 5(a), the maximum emission peak wavelength from the substituted Irpiq, which has a variety of substituent on the phenyl ring can be foreseen. | ||
| Fig. 4 Oxidation potential versus Hammett constant for substituted 2-phenylisoqunoline Ir complexes shown in Table 3. | ||
 | ||
| Fig. 5 (a) Emission energy versus the Hammett constant for substituted 2-phenylisoqunoline Ir complexes shown in Table 3; (b) effect of the Hammett constant on the emission energy. | ||
(4) The mixing of excited states
In the case of Irpiq (1), the lowest excited state was assigned to 3MLCT.10 An analogous argument is used to assign the excited states for the complexes 2–8. Short lifetime, broad luminescence spectra32 and similar absorption peaks33,34 of those complexes show clearly that the lowest triplet excited states of those complexes are predominantly 3MLCT as same as that of Irpiq (1).10,35,36 For more detail, we should consider degree of mixing to understand the photochemical differences of those complexes according to eqn. (3-1).| ΦT = aΦT(ππ*) + bΦT(MLCT) | (3-1) |
Four typical characteristics regarding the emission spectrum and the absorption spectrum of the 3π–π* state were proposed. The first is the vibrational sideband pattern of the luminescence from the 3π–π* state. On the contrary, the broad shape of luminescence spectrum is a typical characteristic of the emission from 3MLCT excited state.33,39 Second is the smaller radiative decay rate constant of 3π–π* state compared to 3MLCT state. Therefore, for the third, the molar absorption constant of the singlet–triplet absorption band of the 3π–π* state is smaller than that of the 3MLCT excited state.24,39 The fourth is the short phosphorescence lifetime for the 3MLCT. The phosphorescent lifetime of the 3MLCT dominant complex is shorter than that of the 3π–π* dominant complex.9,10,40,41
We examined the photophysical properties of complexes 1–8 in view of the above four criteria.
(i) The vibrational sideband patterns of the photoluminescence for the complexes 2, 3 and 4 are more prominent than those of the complexes 1, 5, 6, 7 and 8 as shown in Fig. 6(a) and (b).
 | ||
| Fig. 6 Emission spectra of Ir complexes 1–8. (a) Complexes 1, 5, 6, 7 and 8 with electron withdrawing substituents; (b) complexes 2, 3 and 4 with electron donating substituents. Vibronic structure of (b) is clearly observed in comparison with (a). | ||
(ii) The radiative decay rate constants of complexes 2, 3 and 4 are smaller than those of complexes 1, 5, 6, 7 and 8 as shown in Table 3.
(iii) Furthermore, the absorption peak of the complexes 2, 3 and 4 were lower than that of the complexes 1, 5, 6, 7 and 8. The molar absorption coefficients at the peaks around 590 nm of the former complexes (those are considered as singlet–triplet absorption peaks from ground singlet state to 3MLCT33,36,38) were smaller than that of the latter complexes. The molar absorption coefficients of the complexes 1, 5, 6, 7 and 8 ranges between 1000 and 2000 M−1 cm−1. On the contrary, the molar absorption coefficients of the complexes 2, 3 and 4 are invisible as shown in Table 3.
(iv) Phosphorescent lifetimes of the complexes 1, 5, 6, 7 and 8 were obviously shorter than those of the complexes 2, 3 and 4 as seen in Table 3.
From the above results (the differences of vibrational structure, kr, molar absorption coefficient of singlet–triplet absorption peaks and phosphorescent lifetime), it can be concluded that the complexes 2, 3 and 4 (which have large Hammett constants substituents) have excited states with large contributions of 3π–π* in comparison with the complexes 1, 5, 6, 7 and 8 (which have small Hammett constant substituents).
It is probable that increased electron population of phenyl ring where the HOMO is located enhances the contribution of 3π–π* in the lowest triplet excited state in the iridium complexes. And then, the behaviour of the charge transfer from iridium to ligand decreases.
These results agree with the B3LYP density functional theory (DFT) calculations14 through Mulliken charge difference on iridium atom between the ground state and the lowest triplet excited state. (The charge difference is defined here as a value which charge of the ground state minus charge of the lowest triplet excited state.) The calculations were done for Irppy, Irpiq (1), Ir4F5Mpiq (3) and Ir4Fpiq (2) using Gaussian98 software package (Gaussian, Inc.)24 with a LanL2DZ basis set. Irppy was calculated for reference of well-known material that had the 3MLCT dominant lowest excited state. The calculated Mulliken charge difference between the ground state and the lowest triplet excited state was 0.45 for Irppy, 0.41 for Irpiq (1), 0.33 for Ir4Fpiq (2) and 0.34 for Ir4F5Mpiq (3), respectively. The values of former two complexes show the large differences and the latter two complexes show relatively the small differences. The large difference of the values means large charge transfer from iridium to ligands. The calculation results strongly support the differences between complexes. It is suggested that the 3π–π* mixing of Ir4Fpiq (2) and Ir4F5Mpiq (3), is larger than Irppy and Irpiq (1), owing to the small reduction of Mulliken charge on iridium in the excited state.
(5) Quantum yield and photochemical properties
Table 3 and Fig. 7(a) show the quantum yields of the complexes 1–8. All the complexes have larger quantum yield than Irpiq (1) except Ir5iPrpiq (7) and Ir5Mpiq (8). The quantum yield has a tendency to shift to higher with shortening the maximum emission peak wavelength. | ||
| Fig. 7 Quantum yield and decay rate constants of the complexes. (a) Quantum yield versus emission peak wavelength; (b) radiative decay rate constant versus emission peak wavelength; (c) nonradiative decay rate constant versus emission peakwavelength; (d) plot of ln (knr) versus emission energy gap. | ||
The radiative and nonradiative decay rate constants, knr and kr, are calculated from the phosphorescence yield, ϕ, and the phosphorescence lifetime by following eqn. (3-2) and (3-3).
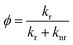 | (3-2) |
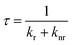 | (3-3) |
From the viewpoint of the relationship between maximum emission peak wavelength of photoluminescent spectrum and decay rate constants, two trends are evident for the substituted iridium 1-phenylisoquinoline derivatives. The nonradiative decay rate constant (knr) increases as the maximum emission peak shift to deep red. Additionally, the radiative decay rate constant (kr) does not show monotonous change by the maximum emission peak shift.
The maximum emission peak wavelength of the complexes with substituent of large Hammett constant is shorter than that of small Hammett constant as shown in Table 3. The energy gaps for complexes 1–8 have been approximated using the maximum emission peak of PL. The plot of ln (knr) versus the energy gap for complexes 1–8 given in Fig. 7(d) shows linear relationship. There is a good qualitative agreement with the “Energy Gap Law”. The Energy Gap Law predicts that the rates of nonradiative decay increase when the energy gap separating the ground and excited state decreases. The relation is based on the vibrational overlap between the ground state and the excited state. And knr is a function of a Franck–Condon overlap integral. In a case of complexes having similar excited states and vibrational coupling, a simplified form of the Energy Gap Law is obtained that predicts a linear relation between ln (knr) and the energy gap.14 The slope of −8.1 eV−1 is similar in magnitude to those found for Os(II), Ru(II)42,43 and Pt(II)17 diimine complexes which have the 3MLCT excited states for the lowest excited triplet states. This correlation suggests that the change of nonradiative decay rate constant is owing to the energy gap of the complexes. Furthermore, this correlation suggests that the charge transfer to the substituted 1-phenylisoquinoline excited states of the all complexes 1, 3–9 have very similar vibrational and electronic components.
Lastly, the radiative decay rate constants of complexes 2–4 are lower than those of complexes 1 and 5–8 due to the changes of the mixing states along the lines of clause (4) [as shown in Fig. 7(b)]. Consequently, complexes 2–4 have larger quantum yield rather than complexes 1 and 5–8.
(6) OLED properties using substituted Irpiq
We have selected Ir4F5Mpiq (3) as a representative material which would show high efficiency and proper red purity for OLED. The OLED was fabricated with Ir4F5Mpiq (3) by the thermal evaporation technique.The evaluation of the device performance was done from the plots of applied voltage versus luminance and power efficiency [Fig. 8(a)] and the plots of applied voltage versus external quantum efficiency [Fig. 8(b)].
 | ||
| Fig. 8 The performance of OLED with Ir4F5Mpiq (3). (a) Power efficiency and luminance versus applied voltage; (b) external quantum efficiency and luminance versus applied voltage for a device in Fig. 1. | ||
The Commission Internationale de L'Eclairage (CIE) system is the standard for evaluating color quality for visual applications.44 The CIE characteristics of the OLEDs with three complexes in the emission layer are shown in Table 5. The device that used Ir4F5Mpiq (3) shows good CIE characteristics, close to the National Television Standards Committee (NTSC) recommended red for a cathode ray tube (CRT). The CIE characteristics of the Ir4F5Mpiq (3) doped OLED are (0.66, 0.34).
It is clear that the EL emission originated from the triplet-excited state of Ir4F5Mpiq (3), because the spectrum of the device was consistent with PL spectrum of the same complex in a dilute toluene solution.
The OLED device showed red electroluminescence with excellent high external quantum efficiency and power efficiency. A maximum external quantum efficiency of 15.5% and a power efficiency of 12.4 lm W−1 were obtained at a luminance 218 cd m−2 and a current density 1.23 mA cm−2. In spite of the gradual decrease of the efficiencies due to triplet–triplet annihilation,45–48 an external quantum efficiency of 7.9% and a power efficiency of 3.7 lm W−1 were obtained at a luminance of 10724 cd m−2 and a current density 119.3 mA cm−2.
Summary
We have synthesized a series of the iridium complexes with 1-phenylisoquinoline as the ligand using various types of substituents. The maximum emission peak wavelength of the Ir complex is tuned from 598 to 635 nm depending on the substituents.The substituents with higher Hammett constants attached to the phenyl ring of 1-phenylisoquinoline caused greater stabilization of the HOMO, which was localized on that phenyl ring. The substituents with the larger Hammett constants stabilized the HOMO, and therefore the t2g orbitals of the iridium, more.
All the complexes discussed here showed 3MLCT predominant mixing states for their lowest excited triplet states. But the degree of the mixing between 3MLCT and 3π–π* states of the lowest excited states varied. The large Hammett constant substituents attached to the phenyl ring enhanced the mixing contribution of 3π–π* excited state due to the reduction of electron density on the iridium. Therefore, we can control the nature of the lowest excited states of substituted Irpiq complexes. And then, we can design the complex and OLED with the objective of desirable emission color and high efficiency.
The OLED device using the Ir complex, Ir4F5Mpiq (3), showed an excellent power efficiency of 12.4 lm W−1, external quantum efficiency of 15.5% and sufficient CIE characteristics (0.66, 0.34) for practical applications.
References
- C. W. Tang and S. A. VanSlyke, Appl. Phys. Lett., 1987, 51, 913 CrossRef CAS.
- M. A. Baldo, D. F. O'Brien, Y. You, A. Shoustikov, S. Sibley, M. E. Thompson and S. R. Forrest, Nature, 1998, 395, 151 CrossRef CAS.
- M. A. Baldo, S. Lamansky, P. E. Burrous, M. E. Thompson and S. R. Forrest, Appl. Phys. Lett., 1999, 75, 4 CrossRef CAS.
- S. Lamansky, P. Djurovich, D. Murphy, F. Abdel-Razzaq, R. Kwong, I. Tsyba, M. Bortz, B. Mui, R. Bau and M. E. Thompson, Inorg. Chem., 2001, 40, 1704 CrossRef CAS.
- J. P. Duan, P. P. Sun and C. H. Cheng, Adv. Mater., 2003, 15, 224 CrossRef CAS.
- A. Beeby, S. Bettington, I. W. Samuel and Z. Wang, J. Mater. Chem., 2003, 13, 80 RSC.
- M. C. DeRosa, P. J. Crutchley and C. E. B. Evans, Inorg. Chem., 2003, 42, 4864 CrossRef CAS.
- J. C. Ostrowski, M. R. Robinson, A. J. Heeger and D. C. Bazan, Chem. Commun., 2002, 784 RSC.
- S. Okada, H. Iwawaki, M. Frugori, J. Kamatani, S. Igawa, T. Moriyama, S. Miura, A. Tsuboyama, T. Takiguchi and H. Mizutani, SID Symp. Dig., ed. J. Morreale, Society for Information Display, San Jose, CA, USA, 2002, p. 1360 Search PubMed.
- A. Tsuboyama, H. Iwawaki, M. Frugori, T. Mukaide, J. Kamatani, S. Igawa, T. Moriyama, S. Miura, T. Takiuchi, S. Okada, M. Hoshino and K. Ueno, J. Am. Chem. Soc., 2003, 125, 12971 CrossRef CAS.
- V. V. Grushin, N. Herron, D. D. LeCloux, W. J. Marshall, V. A. Petrov and Y. Wang, Chem. Commun., 2001, 1494 RSC.
- Y. Wang, N. Herron, V. V. Grushin, D. D. LeCloux and V. A. Petrov, Appl. Phys. Lett., 2001, l79, 449 CrossRef CAS.
- K. Dedeian, P. I. Djurovich, F. O. Garces, G. Carlson and R. J. Watts, Inorg. Chem., 1991, 30, 1685 CrossRef CAS.
- K. Brooks, Y. Babayan, S. Lamansky, P. I. Dojurovich, I. Tsyba, R. Bau and M. E. Thompson, Inorg. Chem., 2002, 41, 3055 CrossRef CAS.
- I. R. Laskar and T. M. Chen, J. Am. Chem. Soc., 2004, 16, 111 CAS.
- W. Zho, C. Liu, L. Su, W. Yang, M. Yuan and Y. Cao, J. Mater. Chem., 2003, 13, 50 RSC.
- S. D. Cummings and R. Eisenberg, J. Am. Chem. Soc., 1996, 118, 1949 CrossRef CAS.
- R. J. Watts and G. A. Crosby, J. Am. Chem. Soc., 1972, 94, 2606 CrossRef CAS.
- Y. J. Su, H. L. Huang, C. L. Li, C. H. Chen, Y. T. Tao, P. T. Chou, S. Datta and R. S. Liu, Adv. Mater., 2003, 15, 884 CrossRef CAS.
- A. Altomare, M. C. Burla, M. Camalli, G. Cascarano, C. Giacovazzo, A. Guagliardi and G. Polidori, J. Appl. Crystallogr., 1994, 27, 435 CrossRef.
- CrystalStructure 3.6.0: Crystal Structure Analysis Package, Rigaku and Rigaku/MSC, The Woodlands, TX, USA, 2000–2004 Search PubMed.
- K. A. King, P. J. Spellane and R. J. Watts, J. Am. Chem. Soc., 1985, 107, 1431 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, V. G. Zakrzewski, J. A. Montgomery, Jr., R. E. Stratmann, J. C. Burant, S. Dapprich, J. M. Millam, A. D. Daniels, K. N. Kudin, M. C. Strain, O. Farkas, J. Tomasi, V. Barone, M. Cossi, R. Cammi, B. Mennucci, C. Pomelli, C. Adamo, S. Clifford, J. Ochterski, G. A. Petersson, P. Y. Ayala, Q. Cui, K. Morokuma, D. K. Malick, A. D. Rabuck, K. Raghavachari, J. B. Foresman, J. Cioslowski, J. V. Ortiz, A. G. Baboul, B. B. Stefanov, G. Liu, A. Liashenko, P. Piskorz, I. Komaromi, R. Gomperts, R. L. Martin, D. J. Fox, T. Keith, M. A. Al-Laham, C. Y. Peng, A. Nanayakkara, C. Gonzalez, M. Challacombe, P. M. W. Gill, B. G. Johnson, W. Chen, M. W. Wong, J. L. Andres, M. Head-Gordon, E. S. Replogle and J. A. Pople, GAUSSIAN 98 (Revision A.9), Gaussian, Inc., Pittsburgh, PA, 1998 Search PubMed.
- A. B. Tamayo, B. D. Alleyne, P. I. Djurovich, S. Lamansky, I. Tsyba, N. N. Ho, R. Bau and M. E. Thompson, J. Am. Chem. Soc., 2003, 125, 7377 CrossRef CAS.
- P. J. Hay and W. R. Wadt, J. Chem. Phys., 1985, 82, 270 CrossRef CAS.
- W. R. Wadt and P. J. Hay, J. Chem. Phys., 1985, 82, 284 CrossRef CAS.
- P. J. Hay and W. R. Wadt, J. Chem. Phys., 1985, 82, 299 CrossRef CAS.
- V. Skarde, M. J. Cook, A. P. Lewis, G. G. McAuliffe, A. J. Thompson and D. J. Robbins, J. Chem. Soc., Perkin Trans. 2, 1984, 1309 RSC.
- C. Hansch, A. Leo, S. H. Unger, K. H. Kim, D. Nikaitani and E. J. Lien, J. Med. Chem., 1973, 16, 1207 CrossRef CAS.
- C. Hansch, S. D. Rockwell, P. Y. C. Jow, A. Leo and E. E. Steller, J. Med. Chem., 1973, 20, 304.
- C. Hansch, A. Leo and R. W. Taft, Chem. Rev., 1991, 91, 165 CrossRef CAS.
- S. Lamansky, P. I. Djurovich, D. Murphy, F. Abdel-Razzaq, H. E. Lee, S. R. Forrest and M. E. Thompson, J. Am. Chem. Soc., 2001, 123, 4304 CrossRef CAS.
- M. G. Colombo, A. Hauser and H. U. Güdel, Inorg. Chem., 1993, 32, 3088 CrossRef CAS.
- M. G. Colombo and H. U. Güdel, Inorg. Chem., 1993, 32, 3081 CrossRef CAS.
- H. Z. Xie, M. W. Liu, O. Y. Wang, X. H. Zhang, C. S. Lee, P. F. Teng, H. L. Kwong, H. Zheng and C. M. Che, Adv. Mater., 2001, 13, 1245 CrossRef CAS.
- M. G. Colombo, T. C. Brunold, T. Riedener, H. U. Güdel, M. Füörtsch and H. B. Bürgi, Inorg. Chem., 1994, 33, 545 CrossRef CAS.
- B. Schmid, F. O. Graces and R. J. Watts, Inorg. Chem., 1994, 33, 9 CrossRef CAS.
- S. Lamansky, P. I. Djurovich, D. Murphy, F. Abdel-Razzaq, H. E. Lee, S. R. Forrest and M. E. Thompson, J. Am. Chem. Soc., 2001, 123, 4304 CrossRef CAS.
- M. G. Colombo and H. U. Güdel, Inorg. Chem., 1993, 32, 3081 CrossRef CAS.
- M. G. Colombo, T. C. Brunold, T. Riedener, H. U. Güdel, M. Füörtsch and H. B. Bürgi, Inorg. Chem., 1994, 33, 545 CrossRef CAS.
- P. Spellane, R. J. Watts and A. Vogler, Inorg. Chem., 1993, 32, 5633 CrossRef CAS.
- J. V. Caspar and T. J. Mayer, J. Am. Chem. Soc, 1983, 105, 5583 CrossRef CAS.
- J. V. Caspar and T. J. Mayer, J. Phys. Chem., 1983, 87, 952 CrossRef CAS.
- J. Whitaker, Electronic Displays; Technology, Design and Applications, McGraw-Hill, New York, 1994, p. 92 Search PubMed.
- C. Adachi, M. A. Baldo and S. R. Forrest, Appl. Phys. Lett., 2000, 77, 904 CrossRef CAS.
- M. A. Baldo and S. R. Forrest, Phys. Rev. B, 62, 10598 Search PubMed.
- M. A. Baldo, C. Adachi and S. R. Forrest, Phys. Rev. B, 62, 10967 Search PubMed.
- C. Adachi, M. A. Baldo, M. E. Thompson and S. R. Forrest, J. Appl. Phys., 2001, 90, 5048 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2005 |