DOI:
10.1039/B416114A
(Paper)
Dalton Trans., 2005, 326-331
Chemistry of phosphine–borane adducts at platinum centers: dehydrocoupling reactivity of Pt(II) dihydrides with P–H bonds
Received 25th October 2004, Accepted 11th November 2004
First published on 7th December 2004
Abstract
The reaction of the Pt(II) dihydride complex cis-[PtH2(dcype)]
(dcype = 1,2-bis(dicyclohexylphosphino)ethane) with the primary or secondary phosphine–borane adducts PhRPH·BH3
(R = H, Ph) was found to exclusively afford the mono-substituted complexes cis-[PtH(PPhR·BH3)(dcype)]
(1: R = H; 2: R = Ph)
via a dehydrocoupling reaction between Pt–H and P–H bonds. Similar reactivity was observed between the uncoordinated phosphines PhRPH (R = H, Ph) and cis-[PtH2(dcype)], which gave cis-[PtH(PPhR)(dcype)]
(4: R = H; 5: R = Ph). The complexes were characterized by 1H, 11B, 13C and 31P NMR spectroscopy, IR, MS and, in the case of 2, X-ray crystallography that confirmed the cis geometries. The di-substituted complex cis-[Pt(PhPH·BH3)2(dcype)]
(3) was prepared from the reaction of cis-[PtCl2(dcype)] with two equivalents of Li[PPhH·BH3]. This suggested that steric reasons alone cannot be used to explain the lack of reactivity with respect to a second dehydrocoupling reaction involving the remaining Pt–H bond in complexes 1, 2, 4 and 5.
Introduction
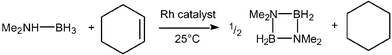
Catalytic dehydrocoupling has recently emerged as a convenient, mild and versatile route for the formation of new bonds between inorganic elements.1 In particular, a range of main group hydride species have been shown to undergo both homo- and heterodehydrocoupling reactions in the presence of a variety of early and late transition metal-catalysts. Catalytic dehydrocoupling reactions to form new Si–Si bonds were first discovered in the mid 1980s.2 Subsequent work has extended this type of method to include, for example, Ge–Ge,3 Sn–Sn,4 P–P,5 Si–P,6 Si–N7 and B–C8 bond forming reactions. Research in our group has focussed on the dehydrocoupling of primary and secondary phosphine–borane adducts (RR′PH·BH3) in the presence of late transition metal-catalysts, which has afforded six- and eight-membered rings [RR′P–BH2]x
(x
= 3, 4), linear oligomeric species RR′PH–BH2–RR′P–BH3 and high molecular weight poly(phosphinoboranes) [RPH–BH2]n
(eqns. (1) and (2)).9,10 This method was extended to the metal-catalyzed dehydrocoupling of primary and secondary amine–borane adducts (RR′NH·BH3), which has afforded cyclic aminoboranes [RR′N–BH2]2 and borazines [RN–BH]3 under mild reaction conditions (eqns. (3) and (4)).10,11 In addition, a tandem catalytic dehydrocoupling–hydrogenation reaction involving a variety of Rh (pre)catalysts and Me2NH·BH3 as a stoichiometric hydrogen source for the hydrogenation of alkenes at 25 °C has also been recently developed (eqn. (5)).12 Recent comparative work between the two systems has indicated the presence of a homogeneous mechanism for phosphine–borane adducts and a heterogeneous mechanism involving Rh(0) colloids in the case of amine–borane analogs.13 Our attempts to further explore the homogeneous mechanism of phosphine–borane dehydrocoupling involved studies of the chemistry of phosphine–borane adducts at platinum centers.14,15 During this work an unusual reaction between cis-[PtH(PPhH·BH3)(depe)]
(depe = 1,2-bis(diethylphosphino)ethane) and PhPH2·BH3 was observed, which afforded the di-substituted complex cis-[Pt(PPhH·BH3)2(depe)].14b This was formally considered to be a dehydrocoupling reaction involving Pt–H and P–H bonds.| |  | (1) |
| |  | (2) |
| |  | (3) |
| |  | (4) |
| |  | (5) |
To date, there are only a few known examples of “dehydrocoupling type” reactions involving either Pt or Pd hydride complexes. For example, Fink and co-workers have reported the reaction of cis-[PtH2(dcype)] with the disilane H3Si–SiH3 which afforded a mixture of cis-[Pt(SiH2SiH3)2(dcype)] and [Pt(µ-SiH2SiH2)(dcype)]2.16 Puddephatt and coworkers have reported the reaction of the dinuclear complex [Pt2Me6(µ-H)(bu2bpy)2]OTf (bu2bpy = 4,4′-di-tert-butyl-2,2′-bipyridine) with HSPh to form the substituted complex [PtMe3(bu2bpy)(SPh)] and H2.17 Recently, Glueck and co-workers have reported the reaction of the bridging Pd hydride complex [Pd2I2(µ-dppf)(µ-H)(µ-PPh2)]
(dppf = 1,1′-bis(diphenylphosphino)ferrocene) with Ph2PH to yield [PdI(µ-PPh2)(Ph2PH)]2.18 However, more detailed investigations into the dehydrocoupling reactivity of Pt hydrides with main group compounds containing E–H bonds have not been performed. In this paper, we report on our detailed investigations of the reactivity of platinum(II) dihydride complexes with phosphines and phosphine–borane adducts bearing P–H bonds.
Results and discussion
The first Pt(II) hydride complexes (e.g.trans-[PtHCl(PEt3)2]) were discovered by Chatt and Shaw in 1957.19 While Pt(IV) hydride species are relevant with respect to C–H bond activation and Pt-catalyzed hydrosilation reactions, they are less well-known than their Pt(II) counterparts.20 A variety of stable Pt(II) dihydride complexes containing tertiary phosphine ligands have been synthesized with both cis and trans geometries.21 However, steric protection in the form of bulky phosphine ligands is usually required to help stabilize these complexes.22 In addition, the reversible loss of dihydrogen has been observed but can be avoided by exposure of these complexes to a hydrogen atmosphere.23 The complex trans-[PtH2(PMe3)2] is one of the first structurally characterized examples of a mononuclear Pt(II) dihydride complex, which was reported by Trogler and co-workers in 1985.24 Therefore, for their combination of stability and reactivity, platinum(II) dihydride complexes with both cis and trans geometries were chosen for preliminary dehydrocoupling studies.Reaction of trans-[PtH2(PR3)2]
(R =
tBu, Me) with PhPH2·BH3
Our first attempts to explore the reactivity of platinum hydrides with the primary phosphine–borane adduct PhPH2·BH3 were performed using the trans dihydride complexes trans-[PtH2(PR3)2]
(R =
tBu, Me). The reaction of trans-[PtH2(PtBu3)2] with 2 equivalents of PhPH2·BH3 was found to result in partial decomposition of the metal complex with the formation of tBu3P·BH3 after 5 h at 25 °C, in addition to unreacted starting materials. The reaction of trans-[PtH2(PMe3)2] with 2 equivalents of PhPH2·BH3 was performed under an atmosphere of hydrogen, as the dihydride complex has been shown to slowly decompose to eliminate hydrogen under a nitrogen atmosphere.24 However, displacement of the phosphine ligands was again observed with the formation of Me3P·BH3. The formation of R3P·BH3
(R =
tBu, Me) in these reactions likely arises due to the fact that the trialkylphosphines are much stronger bases than PhPH2, and thus will undergo exchange with PhPH2·BH3 to afford the tertiary phosphine–borane adducts. In order to avoid this complication, reactions involving platinum dihydrides containing a chelating bis(phosphine) were investigated, as dissociation from the metal center would be inhibited.Synthesis of cis-[PtH(PhRP·BH3)(dcype)]
(1: R = H; 2: R = Ph)
It is known that platinum dihydride complexes with a cis geometry are prone to hydrogen elimination. However, the use of ligands with bulky substituents can help to stabilize these complexes and prevent decomposition.22 For this reason, the chelating ligand 1,2-bis(dicyclohexylphosphino)ethane (dcype) was employed as the large cyclohexyl groups should ensure sufficient steric protection. The complex cis-[PtH2(dcype)] was readily prepared from the reaction of the corresponding dichloride cis-[PtCl2(dcype)] with 2 equivalents of “superhydride” Li[BEt3H]. It has been previously shown that cis-[PtH2(dcype)] undergoes slow, reversible loss of hydrogen to afford the binuclear complex [(dcype)Pt(µ-H)]2 under a nitrogen atmosphere.23 However, in our case [(dcype)Pt(µ-H)]2 was either not observed, or was formed only in very small quantities (<5%) when the reaction was performed under an atmosphere of nitrogen (1 h, 25 °C). The addition of 1 equivalent of PhPH2·BH3 to a solution of cis-[PtH2(dcype)]
(prepared in situ) was found to result in a rapid colour change from red to yellow along with the formation of gas bubbles. The gas released was determined to be H2, as indicated by a resonance at δ 4.46 ppm (lit. δ 4.5 ppm)5c,25 in the 1H NMR spectrum of the reaction mixture. After 24 h, the 1H, 11B and 31P NMR spectra all indicated the formation of cis-[PtH(PPhH·BH3)(dcype)]
(1)
(eqn. (6)). For example, the 31P{1H} NMR spectrum showed three distinct resonances (Fig. 1(a)), suggesting inequivalency of the phosphorus nuclei in the dcype ligand. A doublet of doublets was observed at δ 77.9 ppm, which is due to the PCy2 group that is arranged trans to the phosphine–borane moiety (JPP
= 264 Hz) and cis to the other PCy2 group (JPP
= 2.6 Hz) with additional 195Pt satellites (JPPt
= 2282 Hz). A second doublet of doublets was observed at δ 66.5 ppm, which is due the PCy2 group that is arranged cis to both the phosphine–borane moiety (JPP
= 15 Hz) and the other PCy2 group (JPP
= 2.6 Hz) with associated 195Pt satellites (JPPt
= 1823 Hz). Finally, a broad doublet was observed at δ
−50.6 ppm, which is due to the phosphine–borane moiety with a large trans coupling to one PCy2 group (JPP
= 266 Hz) and associated Pt satellites (JPPt
= 1887 Hz). The 1H NMR spectrum displayed two key resonances associated with 1. A broad doublet was observed at δ 4.7 ppm, which is due to the PH of the phosphine–borane moiety (JHP
= 323 Hz) while a doublet of doublet of doublets was observed at δ
−2.63 ppm, which is due to the PtH hydrogen atom with coupling to a trans PCy2 group (JHP
= 164 Hz) and cis PCy2 and PPhH·BH3 groups (JHP
= 26 and 13 Hz), with associated Pt satellites (JHPt
= 913 Hz)
(Fig. 1(b)). The 11B NMR spectrum of 1 displayed only a broad signal at δ
−33.6 ppm. The IR spectrum of 1 in CH2Cl2 showed absorptions at 2358, 2197 and 2004 cm−1 due to B–H, P–H and Pt–H stretches, respectively.| |  | (6) |
![Selected NMR spectra of cis-[PtH(PPhH·BH3)(dcype)]
1. (a)
31P{1H} NMR: For Pa: JPPcis
= 2.6 Hz, JPPtrans
= 264 Hz, JPtP
= 2282 Hz. Pb: JPPcis
= 2.6 Hz, JPPcis
= 15 Hz, JPtP
= 1823 Hz. Pc: JPPtrans
= 266 Hz, JPtP
= 1887 Hz. A small amount of unreacted PhPH2·BH3 at ca. δ
−49 ppm overlaps with the signal due to Pc yielding the observed unequal doublet. (b)
1H NMR (hydride region): JHPcis
= 13 and 26 Hz, JHPtrans
= 164 Hz, JPtH
= 913 Hz. The increasing baseline at the left edge of the spectrum is due to the impending intense resonances of the protons in the cyclohexyl groups.](/image/article/2005/DT/b416114a/b416114a-f1.gif) |
| | Fig. 1 Selected NMR spectra of cis-[PtH(PPhH·BH3)(dcype)]
1. (a)
31P{1H} NMR: For Pa: JPPcis
= 2.6 Hz, JPPtrans
= 264 Hz, JPtP
= 2282 Hz. Pb: JPPcis
= 2.6 Hz, JPPcis
= 15 Hz, JPtP
= 1823 Hz. Pc: JPPtrans
= 266 Hz, JPtP
= 1887 Hz. A small amount of unreacted PhPH2·BH3 at ca. δ
−49 ppm overlaps with the signal due to Pc yielding the observed unequal doublet. (b)
1H NMR (hydride region): JHPcis
= 13 and 26 Hz, JHPtrans
= 164 Hz, JPtH
= 913 Hz. The increasing baseline at the left edge of the spectrum is due to the impending intense resonances of the protons in the cyclohexyl groups. | |
The reaction of cis-[PtH2(dcype)] with Ph2PH·BH3 was found to afford the corresponding secondary phosphine–borane complex cis-[PtH(PPh2·BH3)(dcype)]
(2), which has many analogous spectroscopic characteristics to those of 1. For example, the 31P{1H} NMR spectrum again showed three different resonances due to the inequivalent PCy2 groups (δ 77.5 and 67.3 ppm) and the PPh2·BH3 moiety (δ
−5.8 ppm). The hydride region of the 1H NMR spectrum displayed a doublet of doublet of doublets at δ
−1.84 ppm due to coupling with three different phosphorus nuclei, while the 11B NMR spectrum consisted of a broad resonance at δ
−30.5 ppm.
X-Ray quality crystals of 2 were grown from THF–hexanes,26 and the molecular structure is shown in Fig. 2. The geometry around the Pt center is distorted square planar, with the hydride and the phosphine–borane moiety in a cis arrangement. Large angles of 170(2) and 176.10(8)° were observed between the trans substituents (H–Pt–P and P–Pt–P, respectively). The Pt–H bond length was determined to be 1.70(7)
Å, slightly longer than the 1.59(4)
Å found in the analogous complex cis-[PtH(PPh2·BH3)(depe)].14b For the phosphine–borane moiety, Pt–P and P–B bond lengths of 2.332(2) and 1.944(11)
Å were found, respectively. For the chelating phosphine, Pt–P bond lengths of 2.269(2) and 2.312(2)
Å were observed with a P–Pt–P bite angle of 87.12(7)°. The Pt–P bond trans to the hydride ligand was found to be much longer than the Pt–P bond cis to the hydride, which likely arises from the larger trans influence exerted by the hydride ligand compared to the phosphine–borane moiety. Similar bonding behaviour has been observed in the complexes cis-[PtH(PPh2·BH3)(depe)]14b and cis-[PtH{P(O)Ph2}{PPh2(OH)}(PEt3)].27
![Molecular structure of cis-[PtH(PPh2·BH3)(dcype)]
2. Selected bond lengths (Å) and angles (°): Pt(1)–P(1) 2.269(2), Pt(1)–P(2) 2.312(2), Pt(1)–P(3) 2.332(2), Pt(1)–H(1Pt) 1.70(7), P(3)–B(1) 1.944(11), P(1)–C(1) 1.855(8), P(2)–C(2) 1.839(8), P(1)–C(21) 1.861(7), P(2)–C(27) 1.832(7), P(3)–C(3) 1.819(8), P(3)–C(9) 1.823(7), C(1)–C(2) 1.554(10); P(1)–Pt(1)–H(1Pt) 83(2), P(2)–Pt(1)–H(1Pt) 170(2), P(3)–Pt(1)–H(1Pt) 93(2), P(1)–Pt(1)–P(2) 87.12(7), P(1)–Pt(1)–P(3) 176.10(8), P(2)–Pt(1)–P(3) 96.77(8), B(1)–P(3)–Pt(1) 123.7(4).](/image/article/2005/DT/b416114a/b416114a-f2.gif) |
| | Fig. 2 Molecular structure of cis-[PtH(PPh2·BH3)(dcype)]
2. Selected bond lengths (Å) and angles (°): Pt(1)–P(1) 2.269(2), Pt(1)–P(2) 2.312(2), Pt(1)–P(3) 2.332(2), Pt(1)–H(1Pt) 1.70(7), P(3)–B(1) 1.944(11), P(1)–C(1) 1.855(8), P(2)–C(2) 1.839(8), P(1)–C(21) 1.861(7), P(2)–C(27) 1.832(7), P(3)–C(3) 1.819(8), P(3)–C(9) 1.823(7), C(1)–C(2) 1.554(10); P(1)–Pt(1)–H(1Pt) 83(2), P(2)–Pt(1)–H(1Pt) 170(2), P(3)–Pt(1)–H(1Pt) 93(2), P(1)–Pt(1)–P(2) 87.12(7), P(1)–Pt(1)–P(3) 176.10(8), P(2)–Pt(1)–P(3) 96.77(8), B(1)–P(3)–Pt(1) 123.7(4). | |
Synthesis of cis-[Pt(PhPH·BH3)2(dcype)]
(3)
One noteworthy observation for the reactivity of 1 and 2 was that only mono-substituted complexes were formed during the dehydrocoupling reactions. For example, the treatment of 1 with a second equivalent of PhPH2·BH3 was found to result in no further reaction at 25 °C. This reactivity is in contrast to the initial dehydrocoupling reaction observed in which the di-substituted complex cis-[Pt(PPhH·BH3)2(depe)] was formed from the reaction of cis-[PtH(PPhH·BH3)(depe)] with PhPH2·BH3.14b The larger cyclohexyl substituents on the dcype ligand in 1 and 2 might be expected to sterically shield the Pt center and prevent close approach of a second phosphine–borane adduct. This would explain the lack of reactivity towards di-substitution compared to the previously reported depe complex that contains smaller ethyl substituents.14b Contrary to this explanation, it was found that the di-substituted species cis-[Pt(PhPH·BH3)2(dcype)]
(3) resulted from the reaction of cis-[PtCl2(dcype)] with 2 equivalents of Li[PPhH·BH3]
(eqn. (7)). For complex 3, the 31P{1H} NMR spectrum showed the presence of two species with resonances at δ 64.6 and −36.6 ppm for 3′ and δ 65.3 and -46.3 ppm for 3″. These isomers are expected to be a mixture of rac
(R,R and S,S) and meso
(R,S and S,R) diastereomers due to the four different substituents on phosphorus (Pt, H, B and Ph). Based on the previous assignment of cis-[Pt(PPhH·BH3)2(depe)] and the trend in the 31P chemical shifts,14b we can tentatively assign 3′ to be the rac diastereomer and 3″ to be the meso diastereomer. However, structural determination by X-ray crystallography would be required to confirm this assignment. In addition, the complex multiplets observed at δ 64.6 and 65.3 ppm occur as a result of a non-first order AA′XX′ spin system due to the chiral phosphorus centers.14b However, as 3 was synthesized by a salt metathesis reaction and not by a consecutive oxidative-addition/reductive-elimination reaction sequence, steric hindrance can not be completely eliminated as a valid reason to explain the lack of reactivity. For example, the insertion of the Pt center in 1 into the P–H bond of PhPH2·BH3 would likely give an intermediate octahedral Pt(IV) complex [PtH2(PPhH·BH3)2(dcype)], which could reductively eliminate H2 to give 3. However, if this intermediate complex is too sterically hindered, the initial oxidative-addition reaction would not be favoured and only monosubstitution might result. In addition, reactions involving 1 and PhPH2·BH3 may require more forcing conditions that were not investigated during the course of this study.| |  | (7) |
Synthesis of cis-[PtH(PhRP)(dcype)]
(4: R = H; 5: R = Ph)
With the reactivity of cis-[PtH2(dcype)] with primary and secondary phosphine–borane adducts established, the reaction of uncoordinated phosphines bearing P–H bonds was also investigated. The treatment of cis-[PtH2(dcype)] with PhPH2 was found to result in a colour change from red to yellow and the formation of H2 gas. Again, the spectroscopic characteristics were consistent with the formation of cis-[PtH(PhPH)(dcype)]
(4)
(eqn. (8)). For example, the 31P{1H} NMR spectrum showed the presence of three resonances due to three different phosphorus nuclei. The two PCy2 groups displayed resonances at δ 79.3 and 65.5 ppm, while the PPhH group showed a signal at δ
−29.5 ppm with a resolved cis coupling of JPP
= 13 Hz. The 1H coupled 31P NMR spectrum of 4 showed that the latter signal was further split into a doublet of doublets with a large coupling to the PH hydrogen atom (JPH
= 269 Hz). The 1H NMR spectrum showed two key resonances associated with 4. A broad doublet at δ 6.06 ppm due to the PH hydrogen atom was observed, as well as a doublet of doublet of doublets at δ
−2.63 ppm with associated 195Pt satellites, which is due to the PtH hydrogen atom. Similarly, the reaction of cis-[PtH2(dcype)] with Ph2PH was found to result in the formation of cis-[PtH(PPh2)(dcype)]
(5), as evidenced by three 31P NMR resonances at δ 75.8, 61.8 and 13.8 ppm and a 1H NMR hydride resonance at δ
−3.38 ppm, which occurred as the expected doublet of doublet of doublets with associated 195Pt satellites.| |  | (8) |
As the P–H hydrogen substituents in borane-complexed phosphines would be expected to be more acidic those of the corresponding free phosphines, phosphine–borane adducts might be anticipated to undergo more facile reaction with basic transition metal hydrides to form dihydrogen due to this greater inherent polarity difference. However, no difference in reactivity between coordinated and uncoordinated free phosphine was observed in this study, suggesting that the acidity of the P–H hydrogen substituents does not affect the reactivity in this case.
Mechanism for the formation of 1, 2, 4 and 5
The formation of complexes 1, 2, 4 and 5 may be expected to occur by consecutive oxidative-addition/reductive-elimination reactions. For example, the insertion of the Pt center in cis-[PtH2(dcype)] into a P–H bond may give an intermediate octahedral polyhydride complex (e.g.
[Pt(H)3(PPh2)(dcype)] in the case of 5), which could undergo reductive-elimination of H2 to yield a mono-substituted complex. Alternatively, the reductive-elimination of H2 from cis-[PtH2(dcype)] may give [Pt(dcype)], which could undergo oxidative-addition with a P–H bond to afford the mono-substituted complex. Unfortunately, no evidence for any intermediate complexes were observed in the NMR spectra of the reaction mixtures. Notably, Böhm and Brookhart and co-workers have reported the catalytic dehydrocoupling of secondary phosphines using the late transition metal catalyst [Cp*Rh(CH2![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH(SiMe3))2].5c They found that the P–H bonds of two phosphine ligands underwent oxidative-addition at the Rh(I) center to afford a detectable Rh(V) dihydride intermediate [Cp*Rh(H)2(PR2)2]. This intermediate was then observed to reductively eliminate H2 and R2P–PR2 and return to the Rh(I) oxidation state. This type of reaction sequence may parallel the observed reactivity of the phosphine or phosphine–borane species at the Pt center in our case.
CH(SiMe3))2].5c They found that the P–H bonds of two phosphine ligands underwent oxidative-addition at the Rh(I) center to afford a detectable Rh(V) dihydride intermediate [Cp*Rh(H)2(PR2)2]. This intermediate was then observed to reductively eliminate H2 and R2P–PR2 and return to the Rh(I) oxidation state. This type of reaction sequence may parallel the observed reactivity of the phosphine or phosphine–borane species at the Pt center in our case.Summary
The reaction of the dihydride complex cis-[PtH2(dcype)] with primary and secondary phosphine–borane adducts or phosphines has been shown to afford the mono-substituted complexes 1, 2, 4 and 5via dehydrocoupling between the Pt–H and P–H bonds. The formation of di-substituted species were not observed, which may be due to the potentially unfavourable steric hindrance present in a Pt(IV) intermediate. With further development, this dehydrocoupling route may prove to be a general method for the formation of new Pt–P bonds which does not rely on metathesis-type salt elimination reactions.Experimental
General procedures and materials
All reactions and product manipulations were performed under an atmosphere of dry nitrogen using standard Schlenk techniques or in an inert atmosphere glovebox filled with dry nitrogen unless otherwise specified. Hexanes was dried via the Grubb's method28 while THF and CH2Cl2 were dried over Na/benzophenone and CaH2, respectively, and distilled prior to use. Li[BEt3H]
(1.0 M in THF), Na (Aldrich), dcype, Ph2PH, PhPH2
(10 wt% in hexanes)
(Strem Chemicals) were purchased and used as received. Naphthalene (Aldrich) was sublimed prior to use. trans-[PtH2(PtBu3)2],29cis-[PtCl2(PMe3)2],24
(PhCN)2PtCl2,30 Ph2PH·BH39b and PhPH2·BH39b were synthesized by literature procedures.Equipment
NMR spectra were recorded on a Varian Gemini 300 MHz or a Varian Unity 400 MHz spectrometer. Chemical shifts are reported relative to residual protonated solvent peaks (1H, 13C) or external BF3·Et2O (11B) or H3PO4
(31P) standards. NMR spectra were obtained at 300 or 400 MHz (1H), 96 MHz (11B), 75 or 100 MHz (13C) or 121 MHz (31P). Mass spectra were obtained with a VG 70-250S mass spectrometer operating in electron impact (EI) mode. Melting points were performed in sealed capillary tubes and are uncorrected. Infrared spectra were obtained on a Perkin Elmer Spectrum One FT-IR spectrometer using KBr windows.X-Ray structural characterization
Diffraction data were collected on a Nonius Kappa-CCD using graphite-monochromated Mo-Kα radiation (λ
= 0.71073 Å). The data were integrated and scaled using the Denzo-SMN package.31 The structure was solved and refined with the SHELXTL-PC V5.1 software package.32 Refinement was by full-matrix least squares on F2 using all data (negative intensities included). The molecular structure is presented with thermal ellipsoids at a 30% probability level and all hydrogen atoms attached to carbon are omitted for clarity. The hydrogen atoms bonded to carbon were included in calculated positions and treated as riding atoms, while those attached to boron or platinum were located and refined with isotropic thermal parameters.Crystallographic data and summary of data collection and refinement for 2. Empirical formula: C38H62BP3Pt, Mr
= 817.69, T
= 150(1) K, λ
= 0.71073 Å, monoclinic, space group P21/n, crystal size = 0.10 × 0.10 × 0.06 mm, a
= 10.7765(6), b
= 20.0924(12), c
= 17.4561(12)
Å, β
= 100.571(3)°, V
= 3715.5(4)
Å3, Z
= 4, Dc
= 1.462 g cm−3, µ
= 3.931 mm−1, F(000)
= 1672, θ range = 2.58–24.99°, index ranges: −12 ≤
h
≤ 12, −21 ≤
k
≤ 23, −20 ≤
l
≤ 20, reflns. collected = 18649, ind. reflns. = 6408, Rint
= 0.0861, GoF on F2
= 1.031, R1 (I > 2σ(I))
= 0.0478, wR2 (all data)
= 0.1154, peak/hole = 1.609/−2.218 e Å−3.CCDC reference number 249141. See http://www.rsc.org/suppdata/dt/b4/b416114a/ for crystallographic data in CIF or other electronic format.
Synthesis of cis-[PtCl2(dcype)]
To a solution of (PhCN)2PtCl2
(0.369 g, 0.781 mmol) in CH2Cl2
(5 mL), a solution of dcype (0.329 g, 0.778 mmol) in CH2Cl2
(2 mL) was added dropwise at 25 °C. The solution was stirred for 4 h and the volatiles were removed to give cis-[PtCl2(dcype)] as a pale yellow solid. Yield: 0.526 g (98%). 1H NMR (300 MHz, CD2Cl2): δ 2.7 (br, PCH2), 2.34 (m, Cy), 2.2 (br, Cy), 2.0–1.6 (m, Cy), 1.4–1.2 (m, Cy). 31P{1H} NMR (CD2Cl2): δ 64.7 (s, JPPt
= 3574 Hz).Reaction of trans-[PtH2(PtBu3)2] with 2 equiv. PhPH2·BH3
To a solution of trans-[PtH2(PtBu3)2]
(0.054 g, 0.090 mmol) in C6D6 in a 5 mm NMR tube, a solution of PhPH2·BH3
(0.023 g, 0.19 mmol) in C6D6 was added at 25 °C. After 5 h, the 11B and 31P NMR spectra of the reaction mixture showed the presence of unreacted trans-[PtH2(PtBu3)2] and PhPH2·BH3, and also tBu3P·BH3
(δP 58.9 (q, JPB
= 56 Hz), δB
−40.8 (d, JBP
= 56 Hz); lit. δP 58.5 (JPB
= 59 Hz), δB
−40.8 (JBP
= 58 Hz)).33Reaction of trans-[PtH2(PMe3)2] with 2 equiv. PhPH2·BH3
A green solution of Na[naphthalide] was prepared from the reaction of Na (0.178 g, 7.74 mmol) and naphthalene (0.277 g, 2.16 mmol) in THF (7.8 mL) at 25 °C for 1.5 h. Under a H2atmosphere, the above solution of Na[naphthalide]
(3.6 mL, ca. 1.0 mmol) was added to a solution of cis-[PtCl2(PMe3)2]
(0.208 g, 0.497 mmol) in THF (15 mL) at 0 °C. The mixture was stirred for 30 min, then warmed to 25 °C to give a brown solution of trans-[PtH2(PMe3)2]. To this solution, PhPH2·BH3
(0.123 g, 0.992 mmol) in THF (3 mL) was added. After 3.5 h, the 11B and 31P NMR spectra of the reaction mixture showed the presence of Me3P·BH3
(δP
−1.1 (q, JPB
= 60 Hz), δB
−36.9 (d, JBP
= 59 Hz); lit. δP
−1.8 (JPB
= 59.8 Hz), δB
−36.0 (JBP
= 59.8 Hz)).34Synthesis of cis-[(dcype)PtH(PPhH·BH3)]
(1)26
In a 5 mm NMR tube, cis-[PtCl2(dcype)]
(0.039 g, 0.057 mmol) was suspended in C6D6, and a solution of Li[BEt3H] in THF (0.11 mL, 0.11 mmol) was added via syringe. After 1 h at 25 °C, the formation of cis-[PtH2(dcype)] was complete as indicated by 31P{1H} NMR: δ 77.2 (s, JPPt
= 1875 Hz); lit. δ 78.2 (s, JPPt
= 1822 Hz).23 A solution of PhPH2·BH3
(0.007 g, 0.06 mmol) in C6D6 was added via syringe and the formation of bubbles were observed. The initial orange–red solution turned yellow in colour after 24 h at 25 °C. The solution was filtered and the volatiles were removed in vacuo. The residue was washed with hexanes (4 × 10 mL), and the residual solvent removed to give 1 as a yellow solid. Crude yield: 0.020 g (48%). Attempts at recrystallization from THF–hexanes by vapour diffusion first afforded a dark yellow oil in which pale yellow crystals of 1 were embedded and could not be cleanly separated. The crystals were determined to be ca. 95% pure by 1H NMR. Attempts at recrystallization by other methods (slow evaporation, solvent layering, cooling saturated solutions) all resulted in the precipitation of either impure powders or oils. 1H NMR (400 MHz, CD2Cl2): δ 7.84 (m, Ph), 7.70 (m, Ph), 4.7 (d br, JHP
= 323 Hz, PH), 2.0–1.5 (m, PCH2 and Cy), 1.4–1.0 (m, Cy), −2.63 (ddd, JHPtrans
= 164 Hz, JHPcis
= 26 Hz, JHPcis
= 13 Hz, JHPt
= 913 Hz, PtH). 11B{1H} NMR (C6D6): δ
−33.6 (br). 13C{1H} NMR (125 MHz, CD2Cl2): δ 135.9 (Ph), 129.5 (Ph), 128.2 (d, JCP
= 8.8 Hz, Ph), 36.5–35.8 (PCH2), 29.6 (m, Cy), 27.6–27.0 (m, Cy), 26.6–26.2 (m, Cy). 31P{1H} NMR (C6D6): δ 77.9 (dd, JPPcis
= 2.6 Hz, JPPtrans
= 264 Hz, JPPt
= 2282 Hz, PCy2), 66.5 (dd, JPPcis
= 2.6 Hz, JPPcis
= 15 Hz, JPPt
= 1823 Hz, PCy2), −50.6 (d br, JPPtrans
= 266 Hz, JPPt
= 1887 Hz, PHPh). IR (CH2Cl2): 2358 (νBH), 2197 (νPH), 2004 (νPtH) cm−1. EI-MS (70 eV): m/z 725 (M+
− BH3
− 2H, 3%).Synthesis of cis-[(dcype)PtH(PPh2·BH3)]
(2)26
Complex 2 was prepared by a procedure similar to 1 using cis-[PtCl2(dcype)]
(0.039 g, 0.057 mmol), Li[BEt3H] in THF (0.11 mL, 0.11 mmol) and Ph2PH·BH3
(0.011 g, 0.055 mmol). Crude yield: 0.016 g (32%). Attempts at recrystallization from THF/hexanes by vapour diffusion afforded a brown oil in which colourless, X-ray quality crystals of 2 were embedded and could not be cleanly separated. The crystals were determined to be ca. 97% pure by 1H NMR. Similar to that of 1, all other attempts at recrystallization by different methods (slow evaporation, solvent layering, cooling saturated solutions) resulted in the precipitation of either impure powders or oils. 1H NMR (300 MHz, C6D6): δ 8.31 (m, Ph), 7.21 (m, Ph), 7.05 (m, Ph), 2.59 (m, PCH2), 2.24 (m, PCH2), 1.7 -1.4 (m, Cy), 1.23 (m, Cy), 1.03 (m, Cy), −1.84 (ddd, JHPtrans
= 171 Hz, JHPcis
= 13 Hz, JHPcis
= 6.4 Hz, JHPt
= 961 Hz, PtH). 11B{1H} NMR (C6D6): δ
−30.5 (br). 13C{1H} NMR (125 MHz, CD2Cl2): δ 134.6 (br, ipso-Ph), 128.5 (Ph), 128.1 (Ph), 127.7 (d, JCP
= 8.4 Hz, Ph), 32.2 (Cy), 30.3 (Cy), 28–26 (m, PCH2), 23.2 (Cy), 14.4 (Cy). 31P{1H} NMR (C6D6): δ 77.5 (dd, JPPcis
= 3.2 Hz, JPPtrans
= 266 Hz, JPPt
= 2220 Hz, PCy2), 67.3 (dd, JPPcis
= 3.2 Hz, JPPcis
= 9.7 Hz, JPPt
= 1873 Hz, PCy2), −5.8 (d br, JPPtrans
= 254 Hz, JPPt
= 2121 Hz, PPh2). IR (CH2Cl2): 2358 (νBH), 2032 (νPtH) cm−1. EI-MS (70 eV): m/z 817 (M+, 1%), 803 (M+
− BH3, 2%).Attempted reaction of cis-[PtH2(dcype)] with 2 equiv. of PhPH2·BH3
In a 5 mm NMR tube, cis-[PtCl2(dcype)]
(0.021 g, 0.030 mmol) was dissolved in C6D6, and a solution of Li[BEt3H] in THF (0.06 mL, 0.06 mmol) was added via syringe. After 1 h at 25 °C, a solution of PhPH2·BH3
(0.008 g, 0.06 mmol) in C6D6 was added via syringe. Upon complete conversion to 1
(18 h, 25 °C), a second equivalent of PhPH2·BH3
(0.007 g, 0.06 mmol) in C6D6 was added. After 24 h, the 31P NMR spectrum indicated the presence of 1 and unreacted PhPH2·BH3 with no evidence of any di-substituted species.Synthesis of cis-[(dcype)Pt(PPhH·BH3)2]
(3)
A solution of nBuLi in hexanes (1.05 mL, 1.68 mmol) was added dropwise to a solution of PhPH2·BH3
(0.209 g, 1.69 mmol) in THF (14 mL) cooled to 0 °C. The mixture was warmed to 25 °C, and 3 mL of solution (corresponding to ca. 0.36 mmol of Li[PPhH·BH3]) was removed and added dropwise to a solution of cis-[PtCl2(dcype)]
(0.123 g, 0.179 mmol) in CH2Cl2
(5 mL) at 25 °C. After stirring the mixture for 18 h, the volatiles were removed, and the yellow oily residue was dissolved in CH2Cl2
(10 mL). The solution was filtered and hexanes (40 mL) added to precipitate a solid. The supernatant was decanted and the residual solvent was removed in vacuo to give 3 as a yellow solid. Pale yellow crystals were obtained by slow evaporation of a CH2Cl2–hexanes solution (1 ∶ 1) over 3–4 days at 25 °C. Despite confirming the purity of 3 by 1H NMR spectroscopy, suitable elemental analysis could not be obtained. Yield: 0.096 g (62%). Mp 129–131 °C. IR (Nujol): 2335 (νBH), 2247 (νPH) cm−1. EI-MS (70 eV): m/z 833 (M+
− 2 BH3
− 2H, 4%), 435 (dcypeBH2+, 12%).3′
(rac diastereomer): 1H NMR (CD2Cl2): δ 7.58 (m, Ph), 7.24 (m, Ph), 4.7 (d br, JHP
= 341 Hz, PH), 2.68 (m, PCH2), 2.30 (m, PCH2), 1.9–1.6 (m, Cy), 1.4–1.0 (m, Cy). 11B{1H} NMR (CD2Cl2): δ
−36.7 (br). 13C{1H} NMR (CD2Cl2): δ 135.2 (d, JCP
= 8.4 Hz, Ph), 134.1 (d, JCP
= 34 Hz, ipso-Ph), 129.4 (s, Ph), 128.3 (d, JCP
= 8.3 Hz, Ph), 36.3 (m, PCH2), 33.0 (m, Cy), 30.3 (m, Cy), 27.6–26.8 (m, Cy), 26.6 (s, Cy). 31P{1H} NMR (CD2Cl2): δ 64.6 (m, JAX′
=
JA′X
= 224 Hz, JAX
=
JA′X′
=
−20 Hz, JXX′
= 8.4 Hz, JAA′
= 0 Hz, JPPt
= 2170 Hz, PCy2), −36.6 (d br, JPPtrans
= 244 Hz, JPPt
= 1725 Hz, PHPh).
3′
(meso diastereomer): 1H NMR (CD2Cl2): δ 7.84 (m, Ph), 7.30 (m, Ph), 4.7 (d br, JHP
= 341 Hz, PH), 2.68 (m, PCH2), 2.30 (m, PCH2), 1.9–1.6 (m, Cy), 1.4–1.0 (m, Cy). 11B{1H} NMR (CD2Cl2): δ
−36.7 (br). 13C{1H} NMR (CD2Cl2): δipso-Ph not observed, 135.8 (d, JCP
= 7.6 Hz, Ph), 129.5 (s, Ph), 128.2 (d, JCP
= 8.4 Hz, Ph), 36.2 (m, PCH2), 30.7 (m, Cy), 29.6 (m, Cy), 27.6–26.8 (m, Cy), 26.1 (s, Cy). 31P{1H} NMR (CD2Cl2): δ 65.3 (m, JAX′
=
JA′X
= 223 Hz, JAX
=
JA′X′
=
−20 Hz, JXX′
= 7.0 Hz, JAA′
= 0 Hz, JPPt
= 2135 Hz, PCy2), −46.3 (d br, JPPtrans
= 257 Hz, JPPt
= 1755 Hz, PHPh).
Synthesis of cis-[(dcype)PtH(PPhH)]
(4)26
Complex 4 was prepared by a procedure similar to 1 using cis-[PtCl2(dcype)]
(0.042 g, 0.061 mmol), Li[BEt3H] in THF (0.12 mL, 0.12 mmol) and PhPH2 in hexanes (0.008 g, 0.073 mmol). Crude yield: 0.024 g (55%). Attempts at recrystallization from THF–hexanes by vapour diffusion afforded small clusters of pale yellow microcrystals of 4 embedded in a brown oil which could not be cleanly separated. The crystals were determined to be ca. 93% pure by 1H NMR. All attempts to recrystallize 4 by other methods (e.g. slow evaporation, solvent layering, cooling saturated solutions) did not result in pure samples. Mp 150 °C. 1H NMR (400 MHz, CD2Cl2): δ 7.69 (m, Ph), 7.27 (m, Ph), 6.06 (d br, JHP
= 250 Hz, PH), 2.1–1.5 (m, PCH2 and Cy), 1.4–1.0 (m, Cy), −2.63 (ddd, JHPtrans
= 122 Hz, JHPcis
= 19 Hz, JHPcis
= 9 Hz, JHPt
= 682 Hz, PtH). 13C{1H} NMR (125 MHz, CD2Cl2): δ 128.4 (d, JCP
= 9 Hz, Ph), 37–35 (m, PCH2), 31–30 (m, Cy), 30–29 (m, Cy), 27.5–27 (m, Cy), 26.7–26.3 (m, Cy). 31P{1H} NMR (C6D6): δ 79.3 (d, JPPtrans
= 228 Hz, JPPt
= 2310 Hz, PCy2), 65.5 (d, JPPcis
= 13 Hz, JPPt
= 1806 Hz, PCy2), −29.5 (dd, JPPcis
= 13 Hz, JPPtrans
= 228 Hz, JPPt
= 1642 Hz, PHPh). Selected 31P NMR (C6D6): δ
−29.4 (dd br, JPH
= 269 Hz). IR (Nujol): 2308 (νPH), 2000 (νPtH) cm−1. EI-MS (70 eV): m/z 617 (dcypePt, 23%).Synthesis of cis-[(dcype)PtH(PPh2)]
(5)26
Complex 5 was prepared by a procedure similar to 1 using cis-[PtCl2(dcype)]
(0.040 g, 0.058 mmol), Li[BEt3H] in THF (0.12 mL, 0.12 mmol) and Ph2PH (0.011 g, 0.059 mmol). Crude yield: 0.030 g (64%). Attempts at recrystallization from THF/hexanes by vapour diffusion occasionally produced pale brown crystals of 5 that were embedded in a dark brown oil and could not be cleanly separated. The crystals were determined to be ca. 90% pure by 1H NMR. All other attempts at recrystallization by other methods (e.g. slow evaporation, solvent layering, cooling saturated solutions) did not result in crystalline material, only impure powders or oils. Mp 149–153 °C. 1H NMR (400 MHz, CD2Cl2): δ 7.77 (m, Ph), 7.39 (m, Ph), 7.30 (m, Ph), 2.2–1.7 (m, PCH2 and Cy), 1.5–1.1 (m, Cy), −3.38 (ddd, JHPtrans
= 121 Hz, JHPcis
= 21 Hz, JHPcis
= 7.8 Hz, JHPt
= 656 Hz, PtH). 13C{1H} NMR (125 MHz, CD2Cl2): δ 128.6 (d, JCP
= 11 Hz, Ph), 128.3 (br, Ph), 127.8 (d, JCP
= 9 Hz, Ph), 37–36 (m, PCH2), 30.5–29.5 (m, Cy), 28.9–28.6 (m, Cy), 27.7–27 (m, Cy), 26.8–26.2 (m, Cy). 31P{1H} NMR (C6D6): δ 75.8 (dd, JPPcis
= 5.2 Hz, JPPtrans
= 283 Hz, JPPt
= 2263 Hz, PCy2), 61.8 (d, JPPcis
= 14 Hz, JPPt
= 2004 Hz, PCy2), 13.8 (dd, JPPtrans
= 228 Hz, JPPcis
= 10 Hz, JPPt
= 2006 Hz, PPh2). IR (Nujol): 1997 (νPtH) cm−1. EI-MS (70 eV): m/z 802 (M+
− H, 3%).Acknowledgements
C. A. J. is grateful for a Natural Sciences and Engineering Research Council of Canada (NSERC) scholarship (2002–2004) and I. M. thanks NSERC for a Discovery Grant and the Canadian Government for a Canada Research Chair.References
-
(a) T. D. Tilley, Acc. Chem. Res., 1993, 26, 22 CrossRef CAS;
(b) F. Gauvin, J. F. Harrod and H. G. Woo, Adv. Organomet. Chem., 1998, 42, 363 CAS;
(c) J. A. Reichl and D. H. Berry, Adv. Organomet. Chem., 1998, 43, 197.
-
(a) C. Aitken, J. F. Harrod and E. Samuel, J. Organomet. Chem., 1985, 279, C11 CrossRef CAS;
(b) C. T. Aitken, J. F. Harrod and E. Samuel, J. Am. Chem. Soc., 1986, 108, 4059 CrossRef CAS.
-
(a) C. Aitken, J. F. Harrod, A. Malek and E. Samuel, J. Organomet. Chem., 1988, 349, 285 CrossRef CAS;
(b) N. Choi and M. Tanaka, J. Organomet. Chem., 1998, 564, 81 CrossRef CAS.
-
(a) T. Imori and T. D. Tilley, J. Chem. Soc., Chem. Commun., 1993, 1607 RSC;
(b) T. Imori, V. Lu, H. Cai and T. D. Tilley, J. Am. Chem. Soc., 1995, 117, 9931 CrossRef CAS;
(c) J. R. Babcock and L. R. Sita, J. Am. Chem. Soc., 1996, 118, 12481 CrossRef CAS.
-
(a) N. Etkin, M. C. Fermin and D. W. Stephan, J. Am. Chem. Soc., 1997, 119, 2954 CrossRef CAS;
(b) A. J. Hoskin and D. W. Stephan, Angew. Chem., Int. Ed., 2001, 40, 1865 CrossRef CAS;
(c) V. P. W. Böhm and M. Brookhart, Angew. Chem., Int. Ed., 2001, 40, 4694 CrossRef CAS.
- R. Shu, L. Hao, J. F. Harrod, H. G. Woo and E. Samuel, J. Am. Chem. Soc., 1998, 120, 12988 CrossRef CAS.
-
(a) H. Q. Lui and J. F. Harrod, Organometallics, 1992, 11, 822 CrossRef CAS;
(b) J. He, H. Q. Liu, J. F. Harrod and R. Hynes, Organometallics, 1994, 13, 336 CrossRef CAS.
- H. Chen, S. Schlecht, T. C. Semple and J. F. Hartwig, Science, 2000, 287, 1995 CrossRef CAS.
-
(a) H. Dorn, R. A. Singh, J. A. Massey, A. J. Lough and I. Manners, Angew. Chem., Int. Ed., 1999, 38, 3321 CrossRef CAS;
(b) H. Dorn, R. A. Singh, J. A. Massey, J. M. Nelson, C. A. Jaska, A. J. Lough and I. Manners, J. Am. Chem. Soc., 2000, 122, 6669 CrossRef CAS;
(c) H. Dorn, E. Vejzovic, A. J. Lough and I. Manners, Inorg. Chem., 2001, 40, 4327 CrossRef CAS;
(d) H. Dorn, J. M. Rodezno, B. Brunnhöfer, E. Rivard, J. A. Massey and I. Manners, Macromolecules, 2003, 36, 291 CrossRef CAS.
- C. A. Jaska, A. Bartole-Scott and I. Manners, Dalton Trans., 2003, 4015 RSC.
-
(a) C. A. Jaska, K. Temple, A. J. Lough and I. Manners, Chem. Commun., 2001, 962 RSC;
(b) C. A. Jaska, K. Temple, A. J. Lough and I. Manners, J. Am. Chem. Soc., 2003, 125, 9424 CrossRef CAS.
- C. A. Jaska and I. Manners, J. Am. Chem. Soc., 2004, 126, 2698 CrossRef CAS.
-
(a) C. A. Jaska and I. Manners, J. Am. Chem. Soc., 2004, 126, 1334 CrossRef CAS;
(b) C. A. Jaska and I. Manners, J. Am. Chem. Soc., 2004, 126, 9776 CrossRef CAS.
-
(a) H. Dorn, C. A. Jaska, R. A. Singh, A. J. Lough and I. Manners, Chem. Commun., 2000, 1041 RSC;
(b) C. A. Jaska, H. Dorn, A. J. Lough and I. Manners, Chem. Eur. J., 2003, 9, 271 CrossRef CAS.
- For other examples of transition metal phosphine–borane complexes, see the following: For M–PB complexes:
(a) K. Kubo, I. Kanemitsu, E. Murakami, T. Mizuta, H. Nakazawa and K. Miyoshi, J. Organomet. Chem., 2004, 689, 2425 CrossRef CAS;
(b) J. R. Moncarz, T. J. Brunker, D. S. Glueck, R. D. Sommer and A. L. Rheingold, J. Am. Chem. Soc., 2003, 125, 1180 CrossRef CAS;
(c) U. Vogel, P. Hoemensch, K. C. Schwan, A. Y. Timoshkin and M. Scheer, Chem. Eur. J., 2003, 9, 515 CrossRef CAS;
(d) S. J. Lancaster, A. J. Mountford, D. L. Hughes, M. Schormann and M. Bochmann, J. Organomet. Chem., 2003, 680, 193 CrossRef CAS;
(e) A. C. Gaumont, M. B. Hursthouse, S. J. Coles and J. M. Brown, Chem. Commun., 1999, 63 RSC;
(f) S. Moreton, Inorg. Chim. Acta, 1994, 215, 67 CrossRef CAS;
(g) W. Angerer, W. S. Sheldrick and W. Malisch, Chem. Ber., 1985, 118, 1261 CAS . For M–BP complexes;
(h) T. Yasue, Y. Kawano and M. Shimoi, Angew. Chem., Int. Ed., 2003, 42, 1727 CrossRef CAS;
(i) T. Yasue, Y. Kawano and M. Shimoi, Chem. Lett., 2000, 58 CrossRef CAS;
(j) Y. Kawano, T. Yasue and M. Shimoi, J. Am. Chem. Soc., 1999, 121, 11744 CrossRef CAS;
(k) M. Shimoi, S. Ikubo, Y. Kawano, K. Katoh and H. Ogino, J. Am. Chem. Soc., 1998, 120, 4222 CrossRef CAS;
(l) D. J. Elliot, C. J. Levy, R. J. Puddephatt, D. G. Holah, A. N. Hughes, V. R. Magnuson and I. M. Moser, Inorg. Chem., 1990, 29, 5014 CrossRef.
- M. J. Michalczyk, C. A. Recatto, J. C. Calabrese and M. J. Fink, J. Am. Chem. Soc., 1992, 114, 7955 CrossRef CAS.
- G. S. Hill, J. J. Vittal and R. J. Puddephatt, Organometallics, 1997, 16, 1209 CrossRef CAS.
- M. A. Zhuravel, J. R. Moncarz, D. S. Glueck, K. C. Lam and A. L. Rheingold, Organometallics, 2000, 19, 3447 CrossRef CAS.
- J. Chatt, L. A. Duncanson and B. L. Shaw, Proc. R. Chem. Soc. London, Ser. A, 1957, 343 CAS.
- R. J. Puddephatt, Coord. Chem. Rev., 2001, 219–221, 157 CrossRef CAS.
-
(a) B. L. Shaw and M. F. Uttley, J. Chem. Soc., Chem. Commun., 1974, 918 RSC;
(b) T. Yoshida, T. Yamagata, T. H. Tulip, J. A. Ibers and S. Otsuka, J. Am. Chem. Soc., 1978, 100, 2063 CrossRef CAS;
(c) R. S. Paonessa and W. C. Trogler, J. Am. Chem. Soc., 1982, 104, 1138 CrossRef CAS;
(d) R. G. Goel, W. O. Ogini and R. C. Srivastava, Organometallics, 1982, 1, 819 CrossRef CAS;
(e) H. C. Clark and M. J. Hampden Smith, J. Am. Chem. Soc., 1986, 108, 3829 CrossRef CAS;
(f) D. L. Packett and W. C. Trogler, Inorg. Chem., 1988, 27, 1768 CrossRef CAS.
- C. J. Moulton and B. L. Shaw, J. Chem. Soc., Chem. Commun., 1976, 365 RSC.
- D. J. Schwartz and R. A. Andersen, J. Am. Chem. Soc., 1995, 117, 4014 CrossRef CAS.
- D. L. Packett, C. M. Jensen, R. L. Cowan, C. E. Strouse and W. C. Trogler, Inorg. Chem., 1985, 24, 3578 CrossRef CAS.
- T. Beringhelli, G. D'Alfonso, M. Panigati, P. Mercandelli and A. Sironi, Chem. Eur. J., 2002, 8, 5340 CrossRef CAS.
- C. A. Jaska, A. J. Lough and I. Manners, Acta Crystallogr., Sect. E, 2004, 60, 1653 CrossRef.
- L.-B. Han, N. Choi and M. Tanaka, Organometallics, 1996, 15, 3259 CrossRef CAS.
- A. B. Pangborn, M. A. Giardello, R. H. Grubbs, R. K. Rosen and F. J. Timmers, Organometallics, 1996, 15, 1518 CrossRef CAS.
- R. G. Goel, W. D. Ogini and R. C. Srivastava, Organometallics, 1982, 1, 819 CrossRef CAS.
- G. K. Anderson and M. Lin, Inorg. Synth., 1990, 28, 60 CAS.
- Z. Otwinowski and W. Minor, Methods Enzymol., 1997, 276, 307 CrossRef CAS.
- G. M. Sheldrick, SHELXTL-PC V5.1, Bruker Analytical X-Ray Systems Inc., Madison, WI, 1997 Search PubMed.
- J. A. Baban and B. P. Roberts, J. Chem. Soc., Perkin Trans. 2, 1984, 1717 RSC.
- A. H. Cowley and M. C. Damasco, J. Am. Chem. Soc., 1971, 93, 6815 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2005 |
Click here to see how this site uses Cookies. View our privacy policy here.