Synthesis, characterization and coordination chemistry of a macrocyclic ligand containing arsenic donors
Christopher D.
Carmichael
and
Michael D.
Fryzuk
*
Department of Chemistry, University of British Columbia, 2036 Main Mall, Vancouver, British Columbia, Canada V6T 1Z1. E-mail: fryzuk@chem.ubc.ca
First published on 14th December 2004
Abstract
The preparation and characterization of the macrocyclic diamido-diarsine ligand [As2N2]Li2(1,4-dioxane) (1) (where As2N2 = PhAs(CH2SiMe2NSiMe2CH2)2AsPh) and a series of early transition metal complexes are presented. The complexes [As2N2]MCl2 (M = Zr, 2; Ti, 4) and the complex ([As2N2]Y)2(µ-Cl)2 (5) can be prepared by reaction of 1 with the corresponding THF adduct of the metal halide. The iodide derivative of 2, [As2N2]ZrI2 (3) can be prepared by reaction with iodotrimethylsilane. The lithium complex 1 displays a very long lithium–arsenic bond distance of 3.162(10) Å, and the yttrium complex 5 is the first known complex containing a yttrium–arsenic bond. Reduction of 2, 3 or 4 using C8K or activated magnesium decomposed the complexes in such a manner that the ligand was separated from the metal centre. Indirect evidence suggests this may be due to reduction of arsenic in the ligand in preference to the metal.
Introduction
Phosphine donors are well suited as ancillary ligands to a wide range of metal centres across the Periodic Table.1 In contrast, arsine donors, the next heaviest member of group 15, are much less common.2–4 While phosphorus-based ligands display strong σ-donor and π-acceptor abilities along with a convenient NMR-active nucleus, arsenic ligands are known to be modest σ-donors and π-acceptors,5 and 75As (I = 3/2) is not a useful NMR nucleus due to its quadrupolar nature.6 Thus, it is not surprising that many arsenic ligands display lower reactivity when compared to similar phosphorus based systems,7,8 although there are exceptions.9,10Previously, the synthesis of mixed-donor macrocyclic diamido-diphosphine system, [P2N2] (where P2N2 = PhP(CH2SiMe2NSiMe2CH2)2PPh)11 and its coordination chemistry with the early transition elements, particularly groups 3 and 4, were reported.12,13 While one can fine tune the reactivity patterns by changing the substituents at phosphorus within the macrocycle, an alternative approach was to change the donor atoms from P to As. In this paper, the successful synthesis of the diamido-diarsine macrocycle, [As2N2] (where As2N2 = PhAs(CH2SiMe2NSiMe2CH2)2AsPh) is reported and its coordination chemistry with Zr, Ti and Y is detailed. The results of attempts to prepare dinitrogen complexes of Zr and Ti stabilized by the [As2N2] ligand system are also discussed.
Results and discussion
Synthesis of [As2N2] ligand
Following the lithium template synthesis developed for the phosphorus based [P2N2]Li2(1,4-dioxane)11 ligand, the synthesis of [As2N2]Li2(1,4-dioxane) (1) is detailed in Scheme 1. Phenylarsine is synthesized in 72% yield by the reduction of phenylarsonic acid using zinc dust and hydrochloric acid.14 Updating the procedure to take advantage of modern Schlenk techniques allows for an approximate 20% increase in isolated yield over the original and more recent publications using the same preparation.15 Reaction of two equivalents of LiAsHPh with the disilazane HN(SiMe2CH2Cl)2 leads to formation of the intermediate diarsinosilazane HN(SiMe2CH2AsHPh)2 in virtually quantitative yield. Further reaction of the diarsinosilazane with another equivalent of HN(SiMe2CH2Cl)2 in the presence of 4 equivalents of BuLi leads to a single product by 1H NMR spectroscopy. Addition of 1,4-dioxane to a concentrated toluene solution of the crude product gives 1 as a fine cream coloured powder isolated in 67% yield by precipitation with hexanes. 1 is sparingly soluble in hexanes and the need to remove hexanes soluble impurities from the product contributes to the mediocre yield. | ||
| Scheme 1 | ||
X-Ray quality crystals of 1 can be grown by placing the concentrated toluene solution in a −40 °C freezer overnight, following 1,4-dioxane addition. The solid-state molecular structure of 1 is presented in Fig. 1; crystal data are given in Table 6, and selected bond distances and angles are detailed in Table 1. The structure is a one-dimensional polymer with the 1,4-dioxane bridging the lithium atoms of adjacent molecules in a head to tail fashion. Both arsenic atoms are associated to a single lithium atom in a syn fashion, like that seen in syn-[P2N2]Li2(THF).11 However, unlike syn-[P2N2]Li2(THF) where the distances are equal by symmetry, the Li–As distances in 1 differ by half an angstrom. The bond distance of 2.658(11) Å between Li(1) and As(1) agrees well with other literature Li–As distances.16–19 It is more difficult to describe the interaction between Li(1) and As(2) as a true bonding interaction; the distance of 3.162(10) Å is significantly longer than the longest distance, 2.799 Å, reported in the literature.20 This distance is, however, considerably shorter than the sum of the van der Waals radii for the two atoms (3.67 Å)21 and suggests that there is a weak interaction present in the solid state.
| N(1)–Li(1) | 2.186(13) | N(1)–Li(2) | 1.982(17) |
| N(2)–Li(1) | 2.106(14) | N(2)–Li(2) | 2.029(15) |
| O(1)–Li(1) | 2.080(13) | O(2)–Li(2*) | 1.998(14) |
| As(1)–Li(1) | 2.658(11) | As(2)⋯Li(1) | 3.162(10) |
| Li(1)–N(1)–Li(2) | 74.8(5) | Li(1)–N(2)–Li(2) | 75.6(6) |
| N(1)–Li(1)–N(2) | 99.4(5) | N(1)–Li(2)–N(2) | 109.5(6) |
| O(1)–Li(1)–N(1) | 145.9(7) | O(2*)–Li(2)–N(2) | 130.4(9) |
| C(13)–As(1)–Li(1) | 152.4(4) | ||
![Molecular structure (ORTEP) of [As2N2]Li2(1,4-dioxane)
(1). Only the ipso carbons of phenyl rings attached to arsenic are shown.](/image/article/2005/DT/b415976d/b415976d-f1.gif) | ||
| Fig. 1 Molecular structure (ORTEP) of [As2N2]Li2(1,4-dioxane) (1). Only the ipso carbons of phenyl rings attached to arsenic are shown. | ||
The solid-state structure in 1 discussed above is not observed in solution; the ambient temperature 1H NMR spectrum is more consistent with a C2v symmetric ligand environment, as it displays resonances due to only two types of silyl methyl protons, reflecting “top and bottom” asymmetry. The o-protons of the arsine phenyl ring appear as a distinct multiplet; the CH2 ring protons appear as a pair of AB doublets. A singlet is observed for the dioxane protons, integrating to one equivalent, and it is assumed to coordinate to the lithium not coordinated to arsenic. The observed symmetry suggests that the [As2N2] ligand framework is flexible in solution and undergoes a fluxional process. Variable-temperature NMR experiments performed on 1 (290–190 K) did not show any decoalescence of the phenyl, methylene or silyl methyl resonances. Thus, it is not possible to distinguish between a solution structure with both arsenic donors coordinated to the lithium ions at all observed temperatures, or a structure in which the arsenic donors engage in an associative/dissociative process with Li(1) that is fast on the NMR timescale. The 1H NMR and 7Li spectra of 1 are both consistent with a symmetrical structure in solution identical to that proposed for the syn isomer of [P2N2]Li2(THF).11 Unlike the phosphine system where two different isomers can be isolated, no spectral evidence for anti-[As2N2]Li2(dioxane) is observed.
Coordination chemistry of [As2N2]
The coordination chemistry of the [P2N2] ligand has been extensively explored and documented,12,22–24 particularly with zirconium.25 In order to study structural and reactivity differences between [P2N2] and [As2N2] complexes, the coordination chemistry of [As2N2] with Zr, Ti and Y was investigated.The synthesis of [As2N2]ZrCl2 (2) is outlined in eqn. (1). Reaction of 1 with ZrCl4(THF)2 or ZrCl4(THT)2 (THT = tetrahydrothiophene) in refluxing toluene for 24 h produced the product in 70% yield. At such elevated reaction temperatures the product is not stable and decomposes over a period of days. Unfortunately, reactions performed at lower temperatures or for shorter periods of time did not go to completion.
 | (1) |
X-Ray quality crystals of 2, containing one equivalent of co-crystallized solvent, were grown from a saturated toluene solution; the solid-state molecular structure is presented in Fig. 2; crystal data are given in Table 6, and selected bond distances and angles are in Table 2. Structurally, 2 assumes a distorted trigonal prismatic geometry, with the two trigonal planes described by Cl(1)As(1)N(1) and the crystallographically related plane Cl(1*)As(1*)N(1*); the chlorides are not located in the N(1)Zr(1)N(2) plane but are rotated out of this plane by approximately 45°. The Zr–N and Zr–Cl bond distances compare favourably to those of the known [P2N2]ZrCl2 complex25 and other similar zirconium complexes.26–28 Very few complexes containing structurally characterized zirconium arsenic bonds are known,29–32 and the Zr–As bond length of 2.8812(4) Å in 2 is in agreement with recently published zirconium diarsine complexes.32 A direct result of the longer Zr–As interaction in 2, as compared to the Zr–P distances in [P2N2]ZrCl2 (2.694 and 2.707 Å),25 is the position of the arsine phenyl rings; the angle between the phenyl rings is about 30° larger than any observed in the [P2N2] complexes of zirconium. Like [P2N2]ZrCl2, complex 2 displays a similar C2 twist; however, due to the intrinsically longer Zr–As bond the degree of twist is much more dramatic, with the As–Zr–As “bite angle” smaller by 25°.25 The extreme C2 twist can also be implicated in the position of the chloride ligands. Because there is no electronic preference for the orbitals in a d0 transition metal complex, the deviation from octahedral coordination about the zirconium centre is steric in nature. The chloride ligands may be rotated to reduce steric interactions with the silyl methyl carbons, C(3) and C(3*) in Fig. 2, that due to the extreme C2 twist are much closer to the metal centre than in [P2N2] complexes.
| Zr(1)–N(1) | 2.098(2) | Zr(1)–N(1*) | 2.098(2) |
| Zr(1)–As(1) | 2.8812(4) | Zr(1)–As(1*) | 2.8812(4) |
| Zr(1)–Cl(1) | 2.4810(7) | Zr(1)–Cl(1*) | 2.4810(7) |
| N(1)–Zr(1)–N(1*) | 111.82(13) | As(1)–Zr(1)–As(1*) | 128.809(18) |
| Cl(1)–Zr(1)–Cl(1*) | 84.02(4) | N(1)–Zr(1)–Cl(1) | 133.39(6) |
| N(1)–Zr(1)–Cl(1*) | 98.39(6) | ||
![Molecular structure (ORTEP) of [As2N2]ZrCl2
(2). Only the ipso carbons of phenyl rings attached to arsenic are shown.](/image/article/2005/DT/b415976d/b415976d-f2.gif) | ||
| Fig. 2 Molecular structure (ORTEP) of [As2N2]ZrCl2 (2). Only the ipso carbons of phenyl rings attached to arsenic are shown. | ||
![Molecular structure (ORTEP) of [As2N2]ZrI2
(3). Only the ipso carbons of phenyl rings attached to arsenic are shown.](/image/article/2005/DT/b415976d/b415976d-f3.gif) | ||
| Fig. 3 Molecular structure (ORTEP) of [As2N2]ZrI2 (3). Only the ipso carbons of phenyl rings attached to arsenic are shown. | ||
In a manner like that of the precursor complex 1, the dichloride 2 adopts a C2v structure in solution at ambient temperature; the methylene protons appearing as a pair of AB doublets, and the silyl methyl protons as two singlets. This indicates that the solution structure and solid-state structure differ, and that some fluxional process is occurring such that on the NMR timescale, an averaged structure is observed. One possibility is that the complex undergoes a dynamic “rocking” motion of the disilylamido donors that possesses a twisted structure at its two extremes and passes through a symmetrical intermediate, as shown in Scheme 2. Variable-temperature NMR experiments performed on 2 (290–220 K) were not able to discern decoalescence of any resonances. The low solubility of 2 in toluene did not permit data collection at temperatures lower than 220 K.
 | ||
| Scheme 2 | ||
Work with [P2N2]Hf complexes has shown that the iodide complex can be a more productive starting material than the chloride.33 The synthesis of [As2N2]ZrI2 (3) is outlined in eqn. (2). Reaction of 2 with an excess (10 equiv.) of iodiotrimethylsilane produced the desired product as a pale yellow solid in 55% yield after washing with small amounts of benzene. 1H NMR spectra of these washings showed a large number of silyl methyl resonances, indicating that a portion of 2 decomposed. If the reaction is conducted using a smaller excess of iodotrimethylsilane, resonances of a new material, presumed to be the mixed iodide chloride species [As2N2]Zr(I)Cl, can be observed in the 1H NMR spectrum.
 | (2) |
The diiodide complex 3 was crystallized from a saturated THF solution; the solid-state molecular structure is shown in Fig. 3 with crystal data and selected bond lengths and angles described in Table 6 and Table 3 respectively. Like the dichloride 2, 3 assumes a trigonal prismatic geometry about the zirconium centre; the trigonal planes are described by I(1)As(1)N(1) and I(2)As(2)N(2), and the iodides are rotated out of the zirconium amido plane by about 45°. The Zr–N distances are comparable to 2; however, the Zr–As distances are shorter by approximately 0.05 Å, suggesting that the presence of the iodide ligands makes the zirconium more electronegative. The Zr–I bonds are comparable to other zirconium diiodide complexes.34,353 displays a less extreme C2 twist than 2, the As(1)–Zr(1)–As(2) “bite angle” is 135.7°, but still much more extreme than [P2N2]ZrCl2.25 In solution, 3 assumes C2v symmetry; the methylene protons appearing as a pair of AB doublets, and the silyl methyl protons are observed as a coincidental singlet in d8-THF, or two singlets if the spectrum is acquired in d6-benzene.
| Zr(1)–N(1) | 2.112(3) | Zr(1)–N(2) | 2.090(3) |
| Zr(1)–As(1) | 2.8326(5) | Zr(1)–As(2) | 2.8719(5) |
| Zr(1)–I(1) | 2.8638(5) | Zr(1)–I(2) | 2.9265(4) |
| N(1)–Zr(1)–N(2) | 110.95(12) | As(1)–Zr(1)–As(2) | 135.745(16) |
| I(1)–Zr(1)–I(2) | 83.888(13) | N(1)–Zr(1)–I(2) | 140.02(9) |
| N(1)–Zr(1)–I(1) | 102.45(8) | ||
Reactions of 2 and 3 with various alkylating agents such as methyllithium or benzyl magnesium chloride resulted in decomposition. 1H NMR spectra of the isolated materials showed many silyl methyl environments, multiple methylene resonances, and broad resonances that could be due to N–H protons. This evidence suggests that the ligand decomposes and potentially becomes separated from the metal centre. How this occurs is unknown but it is possible that an open coordination site on zirconium, created by partial dissociation of one or both arsine donors, could promote complex decomposition.
An investigation of titanium coordination chemistry was prompted by the smaller size of titanium compared to zirconium and the possibilities derived from a greater size mismatch between the arsenic donors and the titanium centre. The synthesis of [As2N2]TiCl2 (4), is outlined in eqn. (3). Reaction of 1 with TiCl4(THF)2 in toluene for five days produces the desired product in 84% yield.
 | (3) |
X-Ray quality crystals of 4, containing 2.5 equivalents of co-crystallized solvent, were grown from a saturated benzene solution. The solid-state molecular structure is shown in Fig. 4 with crystal data and selected bond lengths and angles described in Table 6 and Table 4, respectively. The titanium centre adopts a five-coordinate trigonal bipyramidal structure that includes only one titanium arsenic bond. Such a five-coordinate bonding motif has been observed before in the complex anti-[P2N2]TiCl236 and the four-coordinate complexes anti-[P2N2]MCl (M = Al, Ga).37 In those cases, the second phosphine is unable to bind because the lone pair is oriented away from the metal centre. Complexes with only one coordinated neutral donor have not been observed in the syn-[P2N2] system; however, the significant size differential and orbital mismatch between titanium and arsenic are likely rationales for the lack of coordination. Unlike complex 2, where the Zr–As bonds are elongated, the single Ti–As bond length in 4 agrees well with literature values.38–40 The Ti–N and Ti–Cl bond distances compare favourably to those of similar titanium complexes.41–44
| Ti(1)–N(1) | 1.885(4) | Ti(1)–N(2) | 1.935(4) |
| Ti(1)–As(1) | 2.6865(9) | Ti(1)⋯As(2) | 4.186(1) |
| Ti(1)–Cl(1) | 2.3248(13) | Ti(1)–Cl(2) | 2.2960(13) |
| N(1)–Ti(1)–N(2) | 111.04(15) | N(1)–Ti(1)–Cl(1) | 110.44(11) |
| Cl(1)–Ti(1)–Cl(2) | 92.53(5) | N(1)–Ti(1)–Cl(2) | 107.14(12) |
![Molecular structure (ORTEP) of [As2N2]TiCl2
(4). Silyl methyl groups are omitted for clarity and only the ipso carbons of phenyl rings attached to arsenic are shown.](/image/article/2005/DT/b415976d/b415976d-f4.gif) | ||
| Fig. 4 Molecular structure (ORTEP) of [As2N2]TiCl2 (4). Silyl methyl groups are omitted for clarity and only the ipso carbons of phenyl rings attached to arsenic are shown. | ||
In solution, 4 assumes C2v symmetry; the silylmethyl protons appear as a singlet and the methylene protons appear as a pair of AB doublets in the 1H NMR spectrum, indicating the complex is not five-coordinate in solution, but not whether the complex is four-coordinate or six-coordinate. The absence of additional resonances in the spectrum indicates that slow ligand dissociation is not occuring, in contrast to similar six-coordinate and eight-coordinate arsine complexes of titanium.40 Linewidths do not indicate fluxionality at room temperature, but the obvious differences between the solution and solid-state structures prompted a variable-temperature 1H NMR investigation of 4 in the range 290–180 K. Signal broadening and decoalescence indicating a loss of symmetry occurs, however, no low temperature limiting spectrum was observed. If 4 is six-coordinate in solution it is possible that the complex undergoes a fluxional process similar to that described for complex 2 above. Attempts to synthesize various [As2N2]Ti dialkyl complexes such methyl, benzyl and neosilyl from 4 resulted in reduction to Ti(III) or mixtures of unidentifiable products.
Previous work in our group using yttrium and lanthanide metals with the [P2N2] ligand has led to intriguing carbon–carbon bond formation upon reaction of halide precursors with aryllithium reagents.12,45,46 The biphenyldiide complex ([P2N2]Y)2{µ-η6:η6′-(C6H5)2} can be synthesized from the neosilyl complex and benzene or by direct reaction with phenyllithium; the naphthalene and anthracene complexes can be synthesized by reduction of ([P2N2]Y)2(µ-Cl)2 with C8K in the presence of the appropriate polycyclic aromatic compound. Eqn. (4) outlines the synthesis of the halide precursor ([As2N2]Y)2(µ-Cl)2 (5). Reaction of 1 with YCl3(THF)3 in hot toluene produced 5 in 75% yield. Like 2, 5 is not stable at elevated temperatures, however due to the increased solubility of YCl3(THF)3 in toluene the reaction was facilitated at a lower temperature, reducing decomposition of the product.
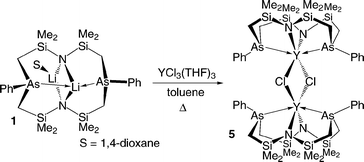 | (4) |
Crystals of 5 suitable for X-ray diffraction experiments were grown from a saturated hexanes solution. The solid-state molecular structure is shown in Fig. 5, with crystal data and selected bond lengths and angles described in Table 6 and Table 5, respectively. Unlike the previously reported ([P2N2]Y)2(µ-Cl)2 structure, where the yttrium centres are structurally different,12 both yttrium centres assume distorted trigonal prismatic geometries; the planes for Y(1) are described by Cl(1)As(1)N(2) and Cl(2)N(1)As(2). The Y–Cl and Y–N bond lengths and the angles of the halide bridge core compare well with those of ([P2N2]Y)2(µ-Cl)2.125 is the first example of a coordination complex containing a Y–As bond.47 The literature contains only five examples of complexes where arsenic is coordinated to any group 3 or lanthanide metal, four complexes of samarium48–50 and one of lutetium.51 As expected, the bond lengths between the smaller yttrium and arsenic are shorter than known lanthanide arsenic bonds. In solution, the molecule behaves in a similar fashion to ([P2N2]Y)2(µ-Cl)2,121H NMR spectra indicate that both ends of the molecule are equivalent, the flexibility of the [As2N2] ligand framework allows the complex to approximate C2v symmetry; all methylene protons appear as a pair of AB doublets and the silyl methyl protons appear as two singlets.
| Y(1)–N(1) | 2.361(4) | Y(1)–N(2) | 2.288(4) |
| Y(1)–As(1) | 2.9644(7) | Y(1)–As(2) | 2.9591(7) |
| Y(2)–N(3) | 2.320(4) | Y(2)–N(4) | 2.283(4) |
| Y(2)–As(3) | 2.9545(7) | Y(2)–As(4) | 2.9968(7) |
| Y(1)–Cl(1) | 2.8119(13) | Y(1)–Cl(2) | 2.7152(13) |
| Y(2)–Cl(1) | 2.7226(13) | Y(2)–Cl(2) | 2.8135(13) |
| Cl(1)–Y(1)–Cl(2) | 78.10(4) | Cl(1)–Y(1)–As(1) | 85.69(3) |
| Cl(1)–Y(1)–As(2) | 133.62(3) | Cl(1)–Y(1)–N(1) | 143.20(12) |
| Cl(1)–Y(1)–N(2) | 98.40(12) | Cl(2)–Y(1)–As(1) | 132.67(4) |
| Cl(2)–Y(1)–As(2) | 83.27(3) | Cl(2)–Y(1)–N(1) | 95.68(11) |
| Cl(2)–Y(1)–N(2) | 142.82(12) | N(1)–Y(1)–N(2) | 107.18(16) |
| As(1)–Y(1)–As(2) | 135.43(2) | Cl(1)–Y(2)–N(4) | 146.28(11) |
| Cl(1)–Y(2)–Cl(2) | 77.95(4) | Cl(1)–Y(2)–As(3) | 124.86(3) |
| Cl(1)–Y(2)–As(4) | 85.00(3) | Cl(1)–Y(2)–N(3) | 97.27(11) |
| Cl(2)–Y(2)–As(4) | 124.61(3) | Cl(2)–Y(2)–As(3) | 85.43(3) |
| Cl(2)–Y(2)–N(4) | 95.05(11) | Cl(2)–Y(2)–N(3) | 150.57(11) |
| As(3)–Y(2)–As(4) | 143.55(2) | N(3)–Y(2)–N(4) | 103.76(15) |
![Molecular structure (ORTEP) of ([As2N2]Y)2(µ-Cl)2
(5). Silyl methyl groups are omitted for clarity, and only the ipso carbons of phenyl rings attached to arsenic are shown.](/image/article/2005/DT/b415976d/b415976d-f5.gif) | ||
| Fig. 5 Molecular structure (ORTEP) of ([As2N2]Y)2(µ-Cl)2 (5). Silyl methyl groups are omitted for clarity, and only the ipso carbons of phenyl rings attached to arsenic are shown. | ||
Reaction of 5 with various alkyl reagents such as Me3SiCH2Li produced a mixture of products from which the expected alkyl could not be cleanly isolated. Attempts to produce and isolate a π-arene complex from 5 have not been successful. Reaction of 5 with C8K in the presence of anthracene yielded intensely blue solutions; however, examination of the isolated solids by 1H NMR showed they were comprised mostly of anthracene and unreacted 5. Small resonances, comprising less than 2% of the sample by integration, were observed that could be tentatively assigned to a biphenyldiide complex based upon the resonances observed for ([P2N2]Y)2{µ-η6:η6′-(C14H10)}.46
Group 4 reduction chemistry
One strategy to prepare early transition metal dinitrogen complexes that has proven useful in the [P2N2] ligand system is to reduce a chloride precursor with an alkali metal reagent, such as potassium graphite (C8K) under dinitrogen.13,52 Reductive pathways to dinitrogen complexes of hafnium have shown that the use of a diiodide precursor can give coordinated dinitrogen complexes where reduction of a dichloride species fails to do so.33,53 If these reductions are performed in the absence of dinitrogen, phosphorus–phenyl activation occurs, giving rise to a highly activated arene bridged dimer.54 Reduction pathways have also been used in the preparation of dinitrogen complexes of titanium.55,56Reaction of 2 or 3 with two equivalents of C8K under dinitrogen did not produce the desired coordinated dinitrogen complex. Reactions conducted under a variety of conditions produced pale hexanes insoluble solids, in contrast to the intense blue color of ([P2N2]Zr)2(µ-η2-N2).13 Mass spectral analysis of the isolated solids did not find any mass peak compatible with a dimeric complex. 1H NMR spectra of the isolated materials showed many silyl methyl environments and multiple methylene resonances, evidence that suggests the ligand decomposes and potentially becomes separated from the metal centre much like that observed for the alkylation reactions described previously. Reductions performed under argon did not yield an activated arene-bridged dimer, only decomposition products were observed, none of which could be assigned to an activated arene bridged dimer. Reductions performed under argon using diphenylacetylene as a trap molecule produced only decomposition products and unreacted diphenylacetylene, suggesting that the decomposition pathway is intramolecular.
The reduction of arsenic(III) to arsenic metal (E° = −0.608 V) occurs at lower potential than the same reduction for phosphorus (E° = −0.87 V) and both are significantly easier to reduce than zirconium (E° = −1.45 V).57 Potassium reducing agents are known to be strongly reducing, with potentials approaching that of free potassium metal (E° = −2.931 V).57 With the possibility that C8K could reduce the arsenic atoms in the macrocycle, milder reducing agents were investigated. The THF adduct of magnesium anthracene is a readily synthesized material with a reduction potential of approximately 2 V.58,59 Reaction of 2 or 3 with one or two equivalents of Mg(anthracene)(THF)3 under a dinitrogen atmosphere produced a dark olive green solid. Examination of these solids by 1H NMR indicated the presence of anthracene, varying amounts of the starting materials and what appeared to be decomposition products. Mass spectral analysis confirmed the absence of any high mass peaks attributable to a dinitrogen complex. Reductions of 2, 3 and 4 using activated magnesium yielded intensely coloured red-brown products; analysis indicated a mixture of products, none of which could be assigned to a dinitrogen complex.
Mass spectral analysis of the products derived from reductions of 2 and 3 did show similarities in the fragmentation patterns. Many fragments could be observed that retained the Si–N bond, the most common fragment observed was [N(SiMe2)2]+. However, no common fragments containing As–C bonds could be assigned. These results suggest that the cleavage of arsenic carbon bonds could be involved in decomposition of the complexes. This also suggests that it may be impossible to cleanly reduce 2 or 3 because of the larger difference in reduction potential between arsenic and zirconium compared to the difference between phosphorus and zirconium.
Conclusions
In this report, the synthesis of a mixed donor diamido-diarsine macrocycle [As2N2], and our attempts to utilize this ligand in dinitrogen activation and aromatic coupling reactions are described. The lithium complex [As2N2]Li2(1,4-dioxane) (1) is a useful reagent to generate the chlorides [As2N2]ZrCl2 (2), [As2N2]TiCl2 (4) and ([As2N2]Y)2(µ-Cl)2 (5) through metathesis type procedures. The related iodide complex [As2N2]ZrI2 (3) can be prepared through the reaction of 2 with iodotrimethylsilane. Attempts to produce coordinated dinitrogen complexes through the reduction of 2, 3 and 4 with C8K, activated magnesium and Mg(anthracene)(THF)3 resulted in decomposition of the parent complex. This same outcome was observed in attempted reduction reactions of the yttrium complex 5 in the presence of polycyclic aromatic hydrocarbons. Evidence links this decomposition to arsenic reduction in the macrocyclic ligand. Attempts to synthesize various alkyl complexes from 2, 3, 4 and 5 resulted in decomposition of the ligand, possibly through an open coordination site on the metal centre created by partial dissociation of one or both arsine donors.Experimental
General procedures
All manipulations were performed under an atmosphere of dry oxygen-free dinitrogen by means of standard Schlenk or glovebox techniques (Vacuum Atmospheres HE-553-2 glovebox equipped with a MO-40-2H purification system and a −40 °C freezer). Hexanes and toluene were purchased anhydrous from Aldrich and further dried by passage through a tower of silica and degassed by passage through a tower of Q-5 catalyst under positive pressure of nitrogen.60 Anhydrous diethyl ether and THF were stored over sieves and distilled from sodium benzophenone ketyl under argon. Nitrogen and argon were dried and deoxygenated by passing the gases through a column containing molecular sieves and MnO. Deuterated benzene was dried by refluxing with molten sodium/potassium alloy in a sealed vessel under partial pressure, then trap-to-trap distilled, and freeze–pump–thaw-degassed three times. Deuterated tetrahydrofuran and toluene were dried by refluxing molten potassium metal in a sealed vessel under vacuum, then trap-to-trap distilled, and freeze–pump–thaw-degassed three times. NMR spectra were recorded on either a Bruker AMX-500 instrument operating at 500.13 MHz for 1H spectra, a Bruker AVA-400 instrument operating at 400.13 MHz for 1H spectra, or a Bruker AVA-300 instrument operating at 300.13 MHz for 1H spectra. 1H NMR spectra were referenced to residual protons in the deuterated solvent and 7Li NMR spectra were referenced to external LiCl (1.0 M in D2O). Elemental analyses were performed by Mr P. Borda or Mr M. Lakha of this department; mass spectra were measured by Mr M. Lapawa, also of this department.Materials
The compounds C8K,61 Mg(anthracene)(THF)3,62 YCl3(THF)3,63 TiCl4(THF)2,64ZrCl4(THF)2,64 and ZrCl4(THT)2,65 were prepared according to literature procedures. Magnesium powder was activated using 1,2-dibromoethane. Phenylarsonic acid and 1.6 M solutions of BuLi in hexanes were purchased from Acros Chemicals and used as received. Iodotrimethylsilane was purchased from Aldrich and used without further purification. All other reagents were obtained from a commercial source and purified by appropriate methods.66Crystallography
In all cases, suitable crystals were selected and mounted on a glass fibre using Paratone-N oil or an acceptable substitute and freezing to −100 °C. All measurements were made on a Rigaku/ADSC CCD area detector with graphite monochromated Mo-Kα radiation. Crystallographic data appear in Table 6. Data was processed using the d*TREK67 module, part of the CrystalClear software package, version 1.3.6 SP0,68 and corrected for Lorentz and polarization effects and absorption. All structures were solved by direct methods using the program SIR97.69 All non-hydrogen atoms were refined anisotropically by least squares procedures on F2 using SHELXL-97.70 Hydrogen atoms were included but not refined; their positional parameters were calculated with fixed C–H bond distances of 0.99 Å for sp2 C, 0.98 Å for sp3 C, and 0.95 Å for aromatic sp C, with Uiso set to 1.2 times the Ueq of the attached sp or sp2 C and 1.5 times the Ueq values of the attached sp3 C atom. Methyl hydrogen torsion angles were determined by electron density. Structure solution and refinements were conducted using the WinGX software package, version 1.64.05.71| [As2N2]Li2(1,4-dioxane) (1) | [As2N2]ZrCl2 (2) | [As2N2]ZrI2 (3) | [As2N2]TiCl2 (4) | ([As2N2]Y)2(µ-Cl)2 (5) | |
|---|---|---|---|---|---|
| CCDC Registry | 250322 | 250323 | 250324 | 250325 | 250326 |
| Formula | C28H50As2Li2N2O2Si4 | C24H42As2Cl2N2Si4Zr·C7H8 | C24H42As2I2N2Si4Zr | C24H42As2Cl2N2Si4Ti·2.5C6H6 | C48H84As4Cl2N4Si8Y2 |
| M | 722.78 | 875.05 | 965.82 | 934.87 | 1490.31 |
| Colour, habit | Colourless, platelet | Colourless, platelet | Colourless, irregular | Orange, irregular | Colourless, irregular |
| Crystal size/mm | 0.20 × 0.10 × 0.05 | 0.30 × 0.20 × 0.05 | 0.25 × 0.10 × 0.05 | 0.3 × 0.2 × 0.1 | 0.2 × 0.15 × 0.15 |
| Crystal system | Orthorhombic | Monoclinic | Monoclinic | Monoclinic | Triclinic |
| Space group | P212121 (no. 19) | C2/c (no. 15) | C2/c (no. 15) | P21/a (no. 16) |
P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) (no. 2)
(no. 2) |
| a/Å | 9.3050(6) | 22.7484(17) | 43.057(2) | 17.8530(13) | 11.5406(9) |
| b/Å | 17.8403(12) | 8.7492(6) | 8.9499(3) | 11.1792(7) | 15.2243(11) |
| c/Å | 21.9501(15) | 21.5246(18) | 19.2595(11) | 23.2490(17) | 20.6305(14) |
| α/° | 90 | 90 | 90 | 90 | 94.8433(19) |
| β/° | 90 | 113.146(2) | 113.8957(15) | 98.286(3) | 98.5004(19) |
| γ/° | 90 | 90 | 90 | 90 | 104.599(2) |
| V/Å3 | 3643.8(3) | 3939.2(5) | 6785.7(6) | 4591.7(6) | 3607.8(4) |
| Z | 4 | 4 | 8 | 4 | 2 |
| T/°C | −100 ± 1 | −100 ± 1 | −100 ± 1 | −100 ± 1 | −100 ± 1 |
| D c/g cm−3 | 1.318 | 1.475 | 1.891 | 1.352 | 1.372 |
| F(000) | 1504 | 1784 | 3744 | 1932 | 1512 |
| µ(Mo-Kα)/mm−1 | 1.992 | 2.227 | 4.239 | 1.866 | 3.659 |
| Transmission factors | 0.7648–1.0000 | 0.7108–1.0000 | 0.6830–1.0000 | 0.7491–1.0000 | 0.8156–1.0000 |
| 2θmax/° | 55.74 | 55.74 | 55.76 | 55.76 | 53.46 |
| Total no. of reflns. | 33235 | 17016 | 29587 | 41258 | 30085 |
| No. of unique reflns. | 7940 | 4353 | 7770 | 10510 | 13428 |
| R merge | 0.0796 | 0.0346 | 0.0353 | 0.0670 | 0.0497 |
| No. reflns. with I ≥ 2σ(I) | 6637 | 3858 | 7226 | 7809 | 11041 |
| No. of variables | 369 | 215 | 324 | 459 | 629 |
| R (F2, all data) | 0.0919 | 0.0432 | 0.0386 | 0.0879 | 0.0668 |
| R w (F2, all data) | 0.1847 | 0.0749 | 0.0905 | 0.1640 | 0.1331 |
| R (F, I > 2σ(I)) | 0.0736 | 0.0357 | 0.0354 | 0.0621 | 0.0530 |
| R w (F, I > 2σ(I)) | 0.1695 | 0.0709 | 0.0883 | 0.1452 | 0.1233 |
| GOF | 1.078 | 1.153 | 1.085 | 1.044 | 1.098 |
CCDC reference numbers 250322–250326.
See http://www.rsc.org/suppdata/dt/b4/b415976d/ for crystallographic data in CIF or other electronic format.
Acknowledgements
Dr B. O. Patrick is gratefully thanked for crystallography assistance and the data collection for complex 2. We thank the NSERC of Canada for generous financial support.References
- J. Emsley and D. Hall, in The Chemistry of Phosphorus, Harper & Row, London, 1976, pp. 177–207 Search PubMed.
- H. Butenschon, Chem. Rev., 2000, 100, 1527–1564 CrossRef CAS.
- O. J. Scherer, Acc. Chem. Res., 1999, 32, 751–762 CrossRef CAS.
- A.-J. DiMaio and A. L. Rheingold, Chem. Rev., 1990, 90, 169–190 CrossRef CAS.
- N. N. Greenwood and A. Earnshaw, in Chemistry of the Elements, Butterworth-Hienemann, Oxford, 2nd edn., 1997, pp. 547–599 Search PubMed.
- R. K. Harris, in NMR and the Periodic Table, ed. R. K. Harris and B. E. Mann, Academic Press, London, 1978, pp. 379–382 Search PubMed.
- J. T. Mague and G. Wilkinson, J. Chem. Soc. A, 1966, 1736–1740 RSC.
- J. T. Carlock, Tetrahedron, 1984, 40, 185–187 CrossRef CAS.
- A. Kojima, C. D. J. Boden and M. Shibasaki, Tetrahedron Lett., 1997, 38, 3459–3460 CrossRef CAS.
- L. A. v. d. Veen, P. K. Keeven, P. C. J. Kamer and P. W. N. M. v. Leeuwen, J. Chem. Soc., Dalton Trans., 2000, 2105–2112 RSC.
- M. D. Fryzuk, J. B. Love and S. J. Rettig, Chem. Commun., 1996, 2783–2784 RSC.
- M. D. Fryzuk, L. Jafarpour, F. M. Kerton, J. B. Love, B. O. Patrick and S. J. Rettig, Organometallics, 2001, 20, 1387–1396 CrossRef CAS.
- M. D. Fryzuk, J. B. Love, S. J. Rettig and V. G. Young, Science, 1997, 275, 1445–1447 CrossRef CAS.
- C. S. Palmer and R. Adams, J. Am. Chem. Soc., 1922, 44, 1356–1382 CrossRef CAS.
- M. A. Beswick, Y. G. Lawson, P. R. Raithby, J. A. Wood and D. S. Wright, J. Chem. Soc., Dalton Trans., 1999, 1921–1922 Search PubMed.
- A. Bashall, A. D. Bond, A. D. Hopkins, S. J. Kidd, M. McPartlin, A. Steiner, R. Wolf, A. D. Woods and D. S. Wright, J. Chem. Soc., Dalton Trans., 2002, 343–351 RSC.
- M. Driess, U. Hoffmanns, S. Martin, K. Merz and H. Pritzkow, Angew. Chem., Int. Ed., 1999, 38, 2733–2736 CrossRef CAS.
- A. Bashall, M. A. Beswick, N. Choi, A. D. Hopkins, S. J. Kidd, Y. G. Lawson, M. E. G. Mosquera, M. McPartlin, P. R. Raithby, A. E. H. Wheatley, J. A. Wood and D. S. Wright, J. Chem. Soc., Dalton Trans., 2000, 479–486 RSC.
- A. Bashall, F. Garcia, A. A. Hopkins, J. A. Wood, M. McPartlin, A. D. Woods and D. S. Wright, Dalton Trans., 2003, 1143–1147 RSC.
- M. Driess, S. Kuntz, K. Merz and H. Pritzkow, Chem. Eur. J., 1998, 4, 1628–1632 CrossRef CAS.
- A. Bondi, J. Phys. Chem., 1964, 68, 441–451 CrossRef CAS.
- M. D. Fryzuk, C. M. Kozak, M. R. Bowdridge, W. Jin, D. Tung, B. O. Patrick and S. J. Rettig, Organometallics, 2001, 20, 3752–3761 CrossRef CAS.
- M. D. Fryzuk, S. A. Johnson, B. O. Patrick, A. Albinati, S. A. Mason and T. F. Koetzle, J. Am. Chem. Soc., 2001, 123, 3960–3973 CrossRef CAS.
- M. D. Fryzuk, G. R. Giesbrecht, S. J. Rettig and G. P. A. Yap, J. Organomet. Chem., 1999, 591, 63–70 CrossRef CAS.
- M. D. Fryzuk, J. B. Love and S. J. Rettig, Organometallics, 1998, 17, 846–853 CrossRef CAS.
- R. R. Schrock, S. W. Seidel, Y. Schrodi and W. M. Davis, Organometallics, 1999, 18, 428–437 CrossRef CAS.
- M. D. Fryzuk, A. Carter and S. J. Rettig, Organometallics, 1992, 11, 469–472 CrossRef CAS.
- M. D. Fryzuk, D. J. Berg and T. S. Haddad, Coord. Chem. Rev., 1990, 99, 137–212 CrossRef CAS.
- E. Hey-Hawkins and F. Lindenberg, Organometallics, 1994, 13, 4643–4644 CrossRef CAS.
- L. Chen and F. A. Cotton, J. Cluster Sci., 1998, 9, 63–91 CrossRef CAS.
- M. Driess, H. Ackermann, J. Aust, K. Merz and C. v. Wullen, Angew. Chem., Int. Ed., 2002, 41, 450–453 CrossRef CAS.
- W. Levason, M. L. Matthews, B. Patel, G. Reid and M. Webster, Dalton Trans., 2004, 3305–3312 RSC.
- M. D. Fryzuk, J. R. Corkin and B. O. Patrick, Can. J. Chem., 2003, 81, 1376–1387 CrossRef CAS.
- Z. Hou, T. L. Breen and D. W. Stephan, Organometallics, 1993, 12, 3158–3167 CrossRef CAS.
- W. A. King, S. D. Bella, A. Gulino, G. Lanza, I. L. Fragala, C. L. Stern and T. J. Marks, J. Am. Chem. Soc., 1999, 121, 355–366 CrossRef CAS.
- G. R. Giesbrecht, PhD Thesis, University of British Columbia, 1998.
- M. D. Fryzuk, G. R. Giesbrecht and S. J. Rettig, Inorg. Chem., 1998, 37, 6928–6934 CrossRef CAS.
- R. A. Jones, S. T. Schwab and B. R. Whittlesey, Polyhedron, 1984, 3, 505–507 CrossRef CAS.
- P. Mercado, A.-J. DiMaio and A. L. Rheingold, Angew. Chem., Int. Ed., 1987, 26, 244–245 CrossRef.
- R. Hart, W. Levason, B. Patel and G. Reid, Eur. J. Inorg. Chem., 2001, 2927–2933 CrossRef.
- G. Jimenez, P. Royo, T. Cuenca and E. Herdtweck, Organometallics, 2002, 21, 2189–2195 CrossRef CAS.
- A. Shafir, M. P. Power, G. D. Whitener and J. Arnold, Organometallics, 2001, 20, 1365–1369 CrossRef CAS.
- R. M. Porter, S. Winston, A. A. Danopoulos and M. B. Hursthouse, J. Chem. Soc., Dalton Trans., 2002, 3290–3299 RSC.
- D. W. Stephan, J. C. Stewart, F. Guerin, S. Courtney, J. Kickham, E. Hollink, C. Beddie, A. Hoskin, T. Graham, P. Wei, R. E. v. H. Spence, W. Xu, L. Koch, X. Gao and D. G. Harrison, Organometallics, 2003, 22, 1937–1947 CrossRef CAS.
- M. D. Fryzuk, J. B. Love and S. J. Rettig, J. Am. Chem. Soc., 1997, 119, 9071–9072 CrossRef CAS.
- M. D. Fryzuk, L. Jafarpour, F. M. Kerton, J. B. Love and S. J. Rettig, Angew. Chem., Int. Ed., 2000, 39, 767–770 CrossRef CAS.
- A search of the Cambridge Crystallographic Data Centre, July 2004 update, did not produce any complexes containing a Y–As bond.
- W. J. Evans, J. T. Leman, J. W. Ziller and S. I. Khan, Inorg. Chem., 1996, 35, 4283–4291 CrossRef CAS.
- F. Nief and L. Ricard, J. Organomet. Chem., 1997, 529, 357–360 CrossRef CAS.
- F. Nief and L. Ricard, Organometallics, 2001, 20, 3884–3890 CrossRef CAS.
- H. Schumann, E. Palamidis, J. Loebel and J. Pickardt, Organometallics, 1988, 7, 1008–1010 CrossRef CAS.
- M. D. Fryzuk, C. M. Kozak, M. R. Bowdridge, B. O. Patrick and S. J. Rettig, J. Am. Chem. Soc., 2002, 124, 8389–8397 CrossRef CAS.
- D. M. Roddick, M. D. Fryzuk, P. F. Seidler, G. L. Hillhouse and J. E. Bercaw, Organometallics, 1985, 4, 97–104 CrossRef CAS.
- M. D. Fryzuk, C. Kozak, P. Mehrkhodavandi, L. Morello, B. O. Patrick and S. J. Rettig, J. Am. Chem. Soc., 2002, 124, 516–517 CrossRef CAS.
- S. M. Mullins, A. P. Duncan, R. G. Bergman and J. Arnold, Inorg. Chem., 2001, 40, 6952–6963 CrossRef CAS.
- T. E. Hanna, I. Keresztes, E. Lobkovsky, W. H. Bernskoetter and P. J. Chirik, Organometallics, 2004, 23, 3448–3458 CrossRef CAS.
- P. Vanysek, in CRC Handbook of Chemistry and Physics, ed. D. R. Lide, CRC Press, Boca Raton, FL, 84th edn. [Online], 2003 Search PubMed.
- B. Bogdanovic, Acc. Chem. Res., 1988, 21, 261–267 CrossRef CAS.
- J. D. Protasiewicz, P. A. Bianconi, I. D. Williams, S. Liu, C. P. Rao and S. J. Lippard, Inorg. Chem., 1992, 31, 4134–4142 CrossRef CAS.
- A. B. Pangborn, M. A. Giardello, R. H. Grubbs, R. K. Rosen and F. J. Timmers, Organometallics, 1996, 15, 1518–1520 CrossRef CAS.
- D. E. Bergbreiter and J. M. Killough, J. Am. Chem. Soc., 1978, 100, 2126–2134 CrossRef CAS.
- B. Bogdanovic, S. Liao, R. Mynott, K. Schlichte and U. Westeppe, Chem. Ber., 1984, 117, 1378–1392 CrossRef.
- K. H. D. Haan, J. L. d. Boer, J. H. Teuben, A. L. Spek, B. Kojic-Prodic, G. R. Hays and R. Huis, Organometallics, 1986, 5, 1726–1733 CrossRef CAS.
- L. E. Manzer, Inorg. Synth., 1982, 21, 135–140 CAS.
- H. Brand and J. Arnold, Organometallics, 1993, 12, 3655–3665 CrossRef CAS.
- D. D. Perrin and W. L. F. Amarego, in Purification of Laboratory Chemicals, Butterworth-Heinemann, Oxford, 3rd edn., 1994 Search PubMed.
- J. W. Pflugrath, Acta Crystallogr., Sect. D, 1999, 55, 1718–1725 CrossRef.
- CrystalClear: An Integrated Program for the Collection and Processing of Area Detector Data, Rigaku Corporation, 2002–2004.
- A. Altomare, M. C. Burla, G. Cammali, M. Cascarano, C. Giacovazzo, A. Guagliardi, A. G. G. Moliterni, G. Polidori and A. Spagna, J. Appl. Crystallogr., 1999, 32, 115–119 CrossRef CAS.
- G. M. Sheldrick, SHELX97: Programs for Crystal Structure Analysis (Release 97-2), University of Göttingen, Germany, 1998 Search PubMed.
- J. Farrugia, J. Appl. Crystallogr., 1999, 32, 837–838 CrossRef.
| This journal is © The Royal Society of Chemistry 2005 |