Selective metal promoted hydrolysis of nitrile groups in the side chain of tetraazamacrocyclic Cu2+-complexes
Liselotte
Siegfried
,
Antonio
Comparone
,
Markus
Neuburger
and
Thomas A.
Kaden
*
Department of Chemistry, Spitalstr. 51, CH-4056, Basel, Switzerland. E-mail: Th.Kaden@unibas.ch
First published on 18th November 2004
Abstract
The metal promoted hydrolysis of nitrile groups in the side chains of tetraazamacrocyclic Cu2+ complexes has been studied by stopped-flow techniques. It is shown that the reaction proceeds by an intramolecular attack of an axially coordinated OH− onto the nitrile group to give the corresponding amide. In alkaline solution the amide then deprotonates and binds to the axial position of the Cu2+ thus preventing further coordination of an OH−. This explains mechanistically that in the Cu2+ complexes of macrocycles carrying two nitrile functions only one is selectively hydrolysed. The nitrile hydrolysis has also been used on a preparative scale to synthesize tetraazamacrocycles with two different side chains. X-Ray diffractions of several products are presented to confirm the structures and the results from the kinetics and equilibria measurements.
Introduction
The functionalisation of azamacrocycles has been developed in the last 20 years to produce a large variety of new ligands.1 By introducing pendant arms with donor groups, chelators have been synthesized, which give metal complexes with new structures and interesting new properties, such as for example contrast agents for NMR-imaging.2 The functionalisation can also be done with the purpose to attach macrocycles through the side chain to a solid support to produce ion exchangers3 or to monoclonal antibodies to give tumour selective diagnostica and therapeutica.4A third type of modification consists in designing side chains with a reactive group so that this group can come close to the metal ion and thus be activated in a similar way as found in metallo enzymes.5 Such low molecular metal complexes have been used for example as models for hydrolases, with the aim to clarify the reaction mechanism and to find structure–reactivity relationships which can only be studied in model compounds by chemical modifications, but not in natural systems. In this way a large amount of information has been gathered.6 Two main reaction pathways have thereby been observed: (a) attack of an external nucleophile onto the coordinated and thus activated group, or (b) attack of an internal coordinated nucleophile onto the organic substrate, which is in close proximity. The differentiation between the two mechanisms is generally easier in the case of inert metal complexes,7 whereas for labile ones it becomes more difficult to prove which reaction path takes place, since in solution rapid equilibria between different species are present. However, if one incorporates such labile metal ions into a macrocyclic ring, one can fix to a certain degree the structure of the species and thus study structure–reactivity relationship in detail.
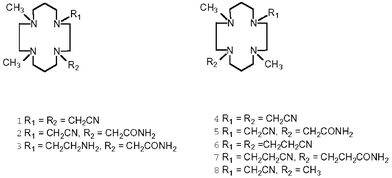
Beside ester, amide and phosphate ester hydrolysis, the reactivity of the nitrile group is also an interesting reaction. Nitrile groups have been incorporated into azamacrocycles and it has been shown that they either point away from the metal ion and do not participate to the metal ion coordination8 or they can bridge to a second metal ion to give polymeric structures in the crystalline state.9 The reactivity of the nitrile group in such complexes was also studied in regard to its methanolysis10 or hydrolysis.9,11 After the observation that in compounds carrying more than one nitrile group a selective reaction with methanol takes place, it was interesting to investigate what would happen in the hydrolysis if more than one nitrile group is present in the macrocyclic complex. We now present a study on the reactivity of the dinitrile derivatives 1, 4, and 6.
Experimental
Materials and methods
1H and 13C NMR spectra were run on a Varian Gemini 300 instrument. δ-values are relative to SiMe4 as internal standard for CDCl3 or to sodium (3-trimethylsilyl)propanesulfonate for D2O solutions. FAB-MS: VG 70-250 with nitrobenzyl alcohol as matrix. Elemental analyses were performed by the analytical laboratory of CIBA AG, Basel. IR spectra were run on a Perkin-Elmer 1600 using KBr pellets.The following compounds were prepared according to the literature: 8,11-dimethyl-1,4,8,11-tetraazacyclotetradecane-1,4-diacetonitrile (1),11 4,11-dimethyl-1,4,8,11-tetraazacyclotetradecane-1,8-diacetonitrile (4),12 4,11-dimethyl-1,4,8,11-tetraazacyclotetradecane-1,8-dipropionitrile (6).12
1,4-Dimethyl-8-(carbamoylmethyl)-1,4,8,11-tetraazacyclotetradecane-11-acetonitrile (2)
In a first test a solution of 1 in DMF (1 ml, 5 × 10−2 M) was mixed with a solution of Cu(ClO4)2·6H2O also in DMF (1 ml, 5 × 10−2 M) and to it a KOH solution (8 ml, 0.1 M) was added. After 1 h the Cu2+ complex was destroyed with KCN (5 ml, 0.1 M) and the aqueous phase extracted with CH2Cl2. The organic phase was dried and chromatographed on a alumina 60 F254 (type E) plastic foil using CHCl3–petroleum ether–MeOH (9 ∶ 1 ∶ 0.5) as solvent. Beside a small spot of the starting compound (Rf = 0.88) the main spot on the chromatogram is the product 2 (Rf = 0.48).Compound 1 (1.0 g, 3.26 mmol) and Cu(ClO4)2·6H2O (1.24 g, 3.27 mmol) were dissolved in DMF (each 3 ml). The two solutions were mixed whereby a blue colour appeared, then NaOH (0.1 M, 10 ml) was added. The mixture was stirred at RT and after 1 h KCN (2.12 g, 32.6 mmol) dissolved in a little water was added, whereby a colour change was observed. The mixture was diluted with water (10 ml) and then extracted with CH2Cl2. The organic phase dried over Na2SO4 was evaporated and the residue taken up with petroleum ether and a few drops CH2Cl2. After filtration the product was precipitated by addition of more petroleum ether. The crude product was recrystallized from petroleum ether. Yield 61%. Mp. 84–86 °C (Found: C, 58.50; H, 10.02; N, 25.54; O, 6.15%. C16H32N6O·0.25 H2O (328.98) requires: C, 58.42; H, 9.96; N, 25.55; O, 6.08%). IR (KBr)/cm−1: 2240 (CN), 1685, 1640 (CONH2). 1H NMR (CDCl3): δ 1.60 (q, 2 CH2CH2CH2), 2.15 (s, 2 NCH3), 2.20–3.0 (m, 8 NCH2CH2), 3.05 (s, NCH2CONH2), 3.60 (s, NCH2CN).
1,4-Dimethyl-8-(carbamoylmethyl)-11-aminoethyl-1,4,8,11-tetraazacyclotetradecane (3)
To 2 (1.0 g, 3.1 mmol) dissolved in abs. EtOH (30 ml), Raney-Ni and liquid NH3 (about 30 ml) were added and the solution was hydrogenated in a Parr reactor at 60 atm during 70 h. After removing the ammonia, the mixture was filtered and evaporated. The oily residue was dissolved in a small quantity of HCl (0.1 M) and so much EtOH added that the hydrochloride precipitated. The product was recrystallized from EtOH–HCl. Yield 1.3 g, 59%. Mp. 226 °C (Found: C, 34.97; H, 8.36; N, 15.24; Cl, 32.16%. C16H41N6OCl5·2.2H2O (550.44) requires: C, 34.91; H, 8.31; N, 15.26; Cl, 32.20%). IR (KBr)/cm−1: 1700, 1630 (CONH2). 1H NMR (D2O): δ 2.1 (q, 2 CH2CH2CH2), 3.0 (s, 2 NCH3), 3.10–3.90 (m, 10 NCH2CH2), 4.0 (s, NCH2CONH2).1,8-Dimethyl-4-(carbamoylmethyl)-1,4,8,11-tetraazacyclotetradecane-11-acetonitrile (5)
The ligand was prepared in analogy to 2 starting from 4. Yield 0.45 g, 43%. Mp. 122–113.5 °C. MS (FAB): 326 (M+ + 2), 325 (M+ + 1), 324 (M+) (Found: C, 59.16; H, 10.19; N, 25.80%. C16H32N6O (324.47) requires: C, 59.23; H, 9.94; N, 25.90%). 1H NMR (CDCl3): δ 1.58, 1.63 (q, 2 CH2CH2CH2), 2.14, 2.19 (s, 2 NCH3), 2.3–2.7 (m, 8 NCH2CH2), 3.05 (s, NCH2CONH2), 3.60 (s, NCH2CN); 13C NMR (CDCl3): δ 24.91, 25.26 (CH2CH2CH2), 41.60 (NCH2), 42.30, 42.88 (NCH3), 50.68, 51.11, 52.66, 53.42, 53.61, 53.80, 55.05, 56.58, 58.40 (NCH2CH2), 114.81 (NCH2CN), 175.89 (NCH2CONH2); IR (KBr)/cm−1: 2226 (CN), 1676, 1611 (CONH2).[1,8-Dimethyl-4-(carbamoylmethyl)-1,4,8,11-tetraazacyclotetradecane-11-acetonitrile]copper(II) diperchlorate, Cu(5)(ClO4)2
To 4 (0.1 g, 0.33 mmol) suspended in water (5 ml) a solution of Cu(ClO4)2·6H2O (0.12 g, 0.32 mmol) in water (10 ml) was added. The mixture turned blue and 4 went into solution by complexation with Cu2+. The solution was left at pH 6–7 for a few days during which cubic crystals formed. Yield 0.02 g, 10%. IR (KBr)/cm−1: 3602, 3382, 2256 (CN), 1678, 1617 (CONH2), 1102 (ClO4−) (Found: C, 33.43; H, 5.51; N, 14.92; Cu, 10.95; Cl, 11.85%. C16H32N6O·0.96Cu(ClO4)2 (576.42) requires: C, 33.34; H, 5.60; N, 14.58; Cu, 10.58; Cl, 11.81%).[1,8-Dimethyl-4-(carbamoylethyl)-1,4,8,11-tetraazacyclotetradecane-11-propionitrile]copper(II) diperchlorate, Cu(7)(ClO4)2
Compound 6 (0.66 g, 1.97 mmol) and Cu(ClO4)2·6H2O (0.74 g, 1.96 mmol) were dissolved in DMF (each 2 ml) and mixed to give a deep blue solution. To it NaOH (0.1 M, 15 ml) was added. The reaction mixture was stirred for 2 h, then the blue precipitate was filtered and dried at high vacuum. Yield 0.6 g, 50%. IR (KBr)/cm−1: 2256 (CN), 1663, 1602 (CONH2), 1098 (ClO4−) (Found: C, 35.14; H, 5.90; N, 13.47; Cu, 10.40; Cl, 11.28%. C18H36N6O·Cu(ClO4)2 (614.97) requires: C 35.16; H 5.90; N 13.67; Cu, 10.33; Cl, 11.53%).Copper(II) complexes of 1,4-dimethyl-8-(carbamoylmethyl)-11-aminoethyl-1,4,8,11-tetraazacyclotetradecane
Measurements
| Cu(5)(ClO4)2·H2O | Cu(7)(ClO4)2 | Cu(3)(ClO4)2·H2O | [Cu(3)]2(ClO4)2.5Cl3.5·C2H5OH·0.75H2O | |
|---|---|---|---|---|
| Formula | C16H34Cl2CuN6O10 | C18H36Cl2CuN6O9 | C16H38Cl2CuN6O10 | C34H81.5Cl6Cu2N12O13.75 |
| M | 604.93 | 614.97 | 608.96 | 1217.40 |
| Crystal system | Monoclinic | Orthorhombic | Triclinic | Monoclinic |
| Space group | P21/c | P212121 |
P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
P21/n |
| a/Å | 12.6801(8) | 9.4996(11) | 13.2769(3) | 21.4405(2) |
| b/Å | 12.7062(14) | 12.3999(15) | 16.8427(3) | 9.6080(1) |
| c/Å | 15.2062(12) | 21.391(2) | 19.9307(4) | 26.1887(3) |
| α/° | 90 | 90 | 105.1318(11) | 90 |
| β/° | 92.868(6) | 90 | 94.3250(9) | 92.7027(7) |
| γ/° | 90 | 90 | 112.9723(11) | 90 |
| Z | 4 | 4 | 6 | 4 |
| V/Å3 | 2446.9(4) | 2519.7(5) | 3881.53(4) | 5388.9(1) |
| D c/kg dm−3 | 1.642 | 1.621 | 1.558 | 1.499 |
| T/K | 173 | 173 | 173 | 173 |
| µ/cm−1 | 1.174 | 1.139 | 1.111 | 1.154 |
| F(000) | 1260 | 1284 | 1902 | 2548 |
| No. of measured refl. | 95325 | 13644 | 32488 | 24391 |
| No. of indep. refl | 8492 | 4394 | 17659 | 12341 |
| No. of refl. in ref. | 5348 | 3319 | 9247 | 7210 |
| No. of variables | 378 | 326 | 973 | 704 |
| Final R value | 0.0287 | 0.0609 | 0.0538 | 0.0386 |
| Final Rw value | 0.0298 | 0.0631 | 0.0539 | 0.0304 |
| R int | 0.09 | 0.15 | 0.02 | 0.02 |
CCDC reference numbers 248566–248569.
See http://www.rsc.org/suppdata/dt/b4/b412857p/ for crystallographic data in CIF or other electronic format.
Results and discussion
Synthetic and structural aspects of the mononitrile monoamide derivatives
If the Cu2+ complexes of the ligands 1, 4, and 6 incorporating two nitrile groups are made alkaline, hydrolysis to the amide occurs, as already observed with analogous mononitrile derivatives.9 The central question is whether both nitrile groups would be hydrolysed to the diamide, or whether only one would react. To check this, the Cu2+ complex of 1 was mixed under conditions typical for the kinetic measurements with 0.1 M KOH, then treated with KCN to destroy the Cu2+ complex of the product, and finally extracted with CH2Cl2. The TLC-chromatogram of the CH2Cl2 extract was run together with the dinitrile starting compound (1). The result was that beside a little spot of the unreacted starting compound the reaction solution contained only the monoamide mononitrile 2.Similar results were obtained when we run reactions for the Cu2+ complexes of 1, 4, and 6 on a preparative scale. The isolated products were always the monoamide mononitriles (Scheme 1). In addition, if one does not destroy the Cu2+ complex with KCN, crystals of good quality for an X-ray diffraction study were obtained for the complexes Cu(5)2+ and Cu(7)2+.
 | ||
| Scheme 1 | ||
The Cu2+ complex of the mononitrile monoamide 5 has the expected structure for a tetra-N-substituted 1,4,8,11-tetraazacyclotetradecane derivative. The macrocycle is in the trans-I configuration,20 in which all four N-substituents are pointing to one side of the N4 plane (Fig. 1) and the Cu2+ ion is coordinated by the four nitrogens of the macrocycle and the carbonyl oxygen of the amide group in the side chain. The exact geometry around the Cu2+ is either distorted trigonal bipyramidal or distorted square pyramidal, the N1–Cu–N3 and N2–Cu–N4 angles being 175.9 and 156.4°, respectively. Cu–N bond lengths are in the normal range (2.03–2.07 Å), whereas the bond length Cu–O (carbonyl) is, at 2.17 Å, somewhat longer than the Cu–N bonds. The side chain with the nitrile group points away from and does not interact with the metal centre. However, it is involved in a hydrogen bond to the NH2-group of the amide of a second molecule, which itself forms an additional hydrogen bond to a water molecule. The net of hydrogen bonds goes even further since the water molecule is also hydrogen bonded to two perchlorates, one of which is disordered. Thus in the crystals a dimeric structure results (Fig. 2), which, however, dissociates in solution into monomers.
 | ||
| Fig. 1 ORTEP plot of the Cu2+ complex with 5. Typical bond lengths (Å): Cu–N(1) 2.063(1), Cu–N(2) 2.063(1), Cu–N(3) 2.078(1), Cu–N(4) 2.033(1), Cu–O(1) 2.174(1). Typical angles (°): N(2)–Cu–N(4) 157.07(6), N(1)–Cu–N(3) 175.58(5), N(1)–Cu–O(1) 79.37(5), N(2)–Cu–O(1) 98.10(5), O(1)–Cu–N(3) 96.37(5), O(1)–Cu–N(4) 104.61(6). | ||
 | ||
| Fig. 2 H-Bond net in the structure of the Cu2+ complex with 5. Typical bond lengths (Å): nitrileN(6)⋯amideH(2) 2.176, amideH(1)⋯waterO(2) 1.997, waterH(4)⋯perchlorateO(5) 1.978, waterH(3)⋯perchlorateO(7) (O(17)) 2.157 (2.042). | ||
The structure of Cu(7)2+ (Fig. 3) closely resembles that of Cu(5)2+. The macrocycle is in the trans-I configuration, the Cu2+, coordinated by the four nitrogens of the macrocycle (Cu–N 2.05–2.14 Å) and the carbonyl oxygen of the amide group (Cu–O 2.14 Å), has a distorted trigonal bipyramidal or square pyramidal structure with angles of 178 and 153° for N(1)–Cu–N(3) and N(2)–Cu–N(4), respectively. Again the nitrile group points away and is not involved in the coordination of the metal ion, but does not form any hydrogen bonds.
 | ||
| Fig. 3 ORTEP plot of the Cu2+ complex with 7. Typical bond lengths (Å): Cu–N(1) 2.105(6), Cu–N(2) 2.066(6), Cu–N(3) 2.143(6), Cu–N(4) 2.046(6), Cu–O(1) 2.143(5). Typical angles (°): N(1)–Cu–N(3) 178.1(2), N(2)–Cu–N(4) 153.1(2), O(1)–Cu–N(1) 89.3(2), O(1)–Cu–N(2) 105.0(2), O(1)–Cu–N(3) 89.3(2), O(1)–Cu–N(4) 101.9(2). | ||
Kinetics of the hydrolysis of the dinitrile derivatives
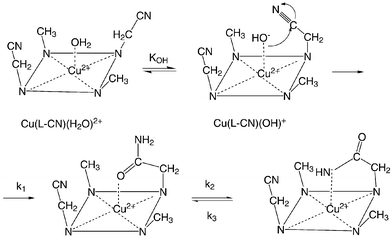
The kinetics of the hydrolysis reaction of the Cu2+ complexes with the dinitriles 1 and 4 in alkaline solution can be fitted with one or two exponentials depending on the wavelength at which the reaction is followed. In general the first and faster step has a relatively small amplitude, whereas the second and slower step exhibits a large amplitude. In this the dinitriles resemble the mononitriles described previously.9 The first step (k1,obs) is the hydrolysis of the nitrile group to the amide and the second step (k2,obs) is the typical change of the O- to the N-coordination of the amide group, which takes place at alkaline pH.The pH dependences of log![[hair space]](https://www.rsc.org/images/entities/char_200a.gif) k1,obs are linear below pH = 12 and show a plateau at higher pH values, indicating that a maximal rate is then reached (Fig. 4). In order to explain these observations we propose Scheme 2.
k1,obs are linear below pH = 12 and show a plateau at higher pH values, indicating that a maximal rate is then reached (Fig. 4). In order to explain these observations we propose Scheme 2.
 | ||
| Fig. 4 pH Dependences of the nitrile hydrolysis (logk1,obs) for the Cu2+ complexes with 1(■ experimental points, — curve calculated with the values of Table 2) and 4
(▲ experimental points, — curve calculated with the values of Table 2). | ||
 | ||
| Scheme 2 | ||
The starting complexes Cu(L–CN)(H2O)2+ are five coordinated species, as indicated by their typical spectra with λmax = 647 and 640 nm for L = 1 and for L = 4, respectively, and are in a fast equilibrium with the hydroxo species Cu(L–CN)(OH)+, in which the OH− group is bound in the axial position. If we assume that Cu(L–CN)(OH)+ is the reactive species we obtain for the rate expression eqn. (1),
| k1, obs = k1KOH[OH−]/(1 + KOH[OH−]) | (1) |
The fast hydrolysis reaction in the two complexes Cu(L–CN)(OH)+ with half-life times of 10–30 ms at pH = 11 results from the very favourable topology of the reactive species (Scheme 2), in which in a five-membered transition state the coordinated OH− group attacks as a nucleophile the nitrile function of the side chain. The role of the metal ion is to organize and orient the reactants so that they are in close vicinity to each other and so that a very effective nucleophilic attack can take place.
The second step (k2,obs) is the deprotonation of the amide group and its interconversion from the O- to the N-coordinated form. The rate constants are pH dependent as expected for such amide deprotonation reactions (Fig. 5) and can be fitted by eqn. (2), where k0 describes the pH independent (eqn. (3)) and k2 the OH− dependent (eqn. (4)) term. The results of the fitting are given in Table 3.
| k2,obs = k0 + k2[OH−] | (2) |
 | (3) |
 | (4) |
 | ||
| Fig. 5 pH dependences of the O- to N-interconversion of the amide group (logk2,obs) for the Cu2+ complex with 2
(▲ experimental points starting from 1, ■ experimental points starting from 2, — curve calculated with the values of Table 3) and 5
(◆ experimental points, — curve calculated with the values of Table 3). | ||
In the case of the Cu2+ complex with 1 we have also studied the O- to N-interconversion starting from the monoamide mononitrile 2 and have obtained the same results as from the hydrolysis experiments of the dinitrile (Fig. 5). In addition also the kinetics of the protonation (k3) of the Cu(L–CONH)+ species described by eqn. (5) was determined.
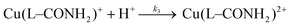 | (5) |
This is an additional proof for the identification of the second step observed during the hydrolysis of the nitrile. It is this second step, which is of paramount importance for the selective hydrolysis of only one of the two nitrile groups. In fact the amide group binds in its deprotonated form to the axial position of the Cu2+ and thus prevents the formation of a new hydroxo complex, which could react with the second nitrile group. In the language of enzymology we would speak of product inhibition.
Cu2+Complexes with the monoamide monoamine derivative 3
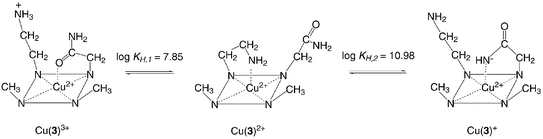
Based on the results of the kinetics of the Cu2+ induced nitrile hydrolysis we have used this selective reaction for synthetic applications to give the mononitrile monoamide derivatives either as Cu2+ complexes or as free ligands (Scheme 1). Generally the syntheses of macrocycles with two different side chains are rather difficult and require selective protection and deprotection with two orthogonal protecting groups. In our case the reaction is simple and selective because of the dual role of the Cu2+ ion, which on one hand activates the hydrolysis according to the mechanism described above, and on the other binds the so formed amide in its deprotonated form, thus inhibiting the further hydrolysis of the second nitrile.Such new products can be used for the preparation of additional disubstituted derivatives since the nitrile group can, for example, be reduced to an amine. In the case of 2 this gives the difunctionalized derivative 3 with an interesting complexation potential. Ligand 3 reacts with Cu2+ to give a 1 ∶ 1 species, which depending on the pH, shows different spectral properties. To quantitatively study the effect of pH on the spectra we have run spectrophotometric pH-titrations of the Cu2+ complex starting from pH ∼ 5, at which the complex is fully formed. Increasing the pH shifts the absorption maximum first from 633 nm to 663 nm, then to 774 nm (Fig. 6). The first change is associated with a protonation constant logKH,1
= 7.85(1), whereas the second one has logKH,2
= 10.98(2) as obtained by fitting the experimental data with the two reactions described by eqns. (6) and (7) with L =
3.
CuLH3+
![[left over right harpoons]](https://www.rsc.org/images/entities/char_21cb.gif) CuL2+
+ H+; KH,1 CuL2+
+ H+; KH,1 | (6) |
| CuL2+
CuLH−1+
+ H+; KH,2 | (7) |
![Spectrophotometric pH titration of the Cu2+ complex with 3 at 25 °C and I
= 0.5 M (KNO3). [CuL2+]
= 2.40 × 10−3 M; (top) original data; (bottom) absorbance as a function of pH at (a)
λ
= 560 nm, (b)
λ
= 740 nm and (c)
λ
= 810 nm. The curves were calculated using the two equilibria described by eqns. (6) and (7) and show the quality of the fitting.](/image/article/2005/DT/b412857p/b412857p-f6.gif) | ||
| Fig. 6 Spectrophotometric pH titration of the Cu2+ complex with 3 at 25 °C and I = 0.5 M (KNO3). [CuL2+] = 2.40 × 10−3 M; (top) original data; (bottom) absorbance as a function of pH at (a) λ = 560 nm, (b) λ = 740 nm and (c) λ = 810 nm. The curves were calculated using the two equilibria described by eqns. (6) and (7) and show the quality of the fitting. | ||
The pH dependent spectral changes in the Cu2+ complex with 3 can be understood by assuming that the side chains of the ligand interact with the axial position of the Cu2+ coordinated by the macrocycle (Scheme 3). In acidic solution the amino group is protonated so that only the amide group can interact with its carbonyl oxygen. Increasing the pH deprotonates the ammonium group to an amine, which being a better donor displaces the amide and binds in the axial position. The shift from 633 to 663 nm is an indication that a stronger donor binds in the axial position. A comparison with the Cu2+ complex of 1,4,8-trimethyl-11-(2-aminoethyl)-1,4,8,11-tetraazacyclotetradecane, in which a shift from 640 to 684 nm is observed,21 when the amino group axially coordinates, shows that a similar reaction also takes place in the Cu2+ complex with 3. Further increase of the pH allows to deprotonate and coordinate the amide group through the nitrogen. Again a shift to longer wavelength is a clear indication that an even stronger donor axially interacts with Cu2+ and replaces the amino group. The spectral properties can be compared to those of the Cu2+ complex with 1,4,8-trimethyl-11-carbamoylmethyl-1,4,8,11-tetraazacyclotetradecane with λmax = 735 nm.21 The equilibria in solution and the structures of the species involved in them are additionally confirmed by the following X-ray diffraction studies.
 | ||
| Scheme 3 | ||
The structure of the Cu2+ complex with 3, which was crystallized at pH ∼ 5, consists of two Cu(3)3+ cations of very similar geometry, of 3.5 Cl−, and 2.5 ClO4− ions per unit cell. In the two cations the Cu2+ is pentacoordinated by the four nitrogens of the macrocycle and the carbonyl oxygen of the amide group (Fig. 7). The amino group of the second side chain is protonated and cannot interact with the metal ion. Cu–N and Cu–O bond lengths are in the normal range and comparable to those of other Cu2+ complexes described in this paper. The macrocycle is as expected in the trans-I conformation.
 | ||
| Fig. 7 ORTEP plot one of the two species of the unit cell of the Cu2+ complex with 3 crystallized at pH 5. Typical bond lengths (Å): Cu(1)–N(1) 2.081(3), Cu(1)–N(2) 2.106(3), Cu(1)–N(3) 2.049(3), Cu(1)–N(4) 2.076(3), Cu(1)–O(1) 2.191(2). Typical angles (°): N(1)–Cu(1)–N(3) 178.11(12), N(2)–Cu(1)–N(4) 153.58(11), O(1)–Cu(1)–N(1) 79.5(1), O(1)–Cu(1)–N(2) 101.1(1), N(3)–Cu(1)–O(1) 98.72(11), N(4)–Cu(1)–O(1) 104.77(10). | ||
When the same complex is crystallized at pH ∼ 9 a new species with a pentacoordinate Cu2+ (three per unit cell) is found. The difference to the previous structure is that now the amino group of the side chain is coordinated to the metal ion and has replaced the carbonyl oxygen of the amide (Fig. 8). Bond lengths Cu–N are in the normal range (2.07–2.12 Å), with a somewhat longer Cu–N bond (2.18–2.21 Å) for the amino group in the side chain. The macrocycle is in the trans-I configuration.20 The amide group is pointing away from the metal ion and is not involved in a coordinative bond.
 | ||
| Fig. 8 ORTEP plot of one of the three species of the unit cell the Cu2+ complex with 3 crystallized at pH 9. Typical bond lengths (Å): Cu(1)–N(1) 2.084(3), Cu(1)–N(2) 2.138(3), Cu(1)–N(3) 2.076(4), Cu(1)–N(4) 2.110(3), Cu(1)–N(5) 2.214(4). Typical angles (°): N(1)–Cu(1)–N(3) 177.18(15), N(2)–Cu(1)–N(4) 147.32(14), N(1)–Cu(1)–N(5) 82.33(14), N(2)–Cu(1)–N(5) 109.37(14), N(3)–Cu(1)–N(5) 100.49(14), N(4)–Cu(1)–N(5) 102.94(14). | ||
Conclusions
The selective hydrolysis of only one nitrile group in the dinitrile derivatives 1, 4 and 6 is possible in the corresponding Cu2+ complexes, since on one hand the Cu2+ ion can axially coordinate an OH− group, which as a nucleophile can attack the nitrile group and on the other can bind the deprotonated amide group which is the end product. Through coordination of the amide nitrogen the hydrolysis of the second nitrile group is inhibited so that the monoamide mononitrile becomes the end product of the reaction. In this case the synthesis of macrocycles with two different side chains is an easy process with no necessity of protection and deprotection steps at all. In addition the monoamide mononitrile derivatives are valuable intermediates for further modification of the side chain. An example of this is given through the reduction of the nitrile group to an amine. Such difunctionalized macrocycles are interesting ligands, since depending on the pH, one or the other group is coordinated. This allows to modify the properties of the metal ion by controlling the pH of the solution.Acknowledgements
This work was supported by the Swiss National Science Foundation (Project N. 2000-66826.01) and this is gratefully acknowledged.References
- (a) Th. A. Kaden, Top. Curr. Chem., 1984, 121, 157 CAS; (b) P. V. Bernhardt and G. A. Lawrance, Coord. Chem. Rev., 1990, 104, 297 CrossRef CAS; (c) M. Meyer, V. Dahaoni-Gindrey, C. Lecomte and R. Guilard, Coord. Chem. Rev., 1998, 178–180, 1313 CrossRef.
- (a) R. B. Lauffer, Chem. Rev., 1987, 87, 901 CrossRef CAS; (b) D. Parker and J. A. G. Williams, J. Chem. Soc., Dalton Trans., 1996, 3613 RSC; (c) P. Caravan, J. J. Ellisom, T. J. McMurry and R. B. Lauffer, Chem. Rev., 1999, 99, 2293 CrossRef CAS.
- (a) S. Blain, P. Appriou, H. Chaumeil and H. Handel, Anal. Chim. Acta, 1990, 232, 331 CrossRef CAS; (b) A. J. Blake, N. R. Champness, S. M. Howdle and P. B. Webb, Inorg. Chem., 2000, 35, 1035 CrossRef CAS.
- D. Parker, Chem. Soc. Rev., 1990, 19, 271 RSC.
- (a) W. Kaim and B. Schwedersk, Bioanorganische Chemie, Treuber Verlag, Stuttgart, 1991 Search PubMed; (b) S. J. Lippard and J. M. Berg, Principles of Bioinorganic Chemistry, University Science Books, Mill Valley, CA, 1994 Search PubMed.
- (a) R. Breslow, R. Fairweather and J. Keana, J. Am. Chem. Soc., 1967, 89, 2135 CrossRef CAS; (b) Y. Gultneh, A. Allwar, B. Ahvazi, D. Blaise, R. J. Butcher and J. Jasinski, Inorg. Chim. Acta, 1996, 241, 31 CrossRef CAS; (c) T. Itoh, H. Hisada, T. Sumiya, M. Hosono, Y. Usui and Y. Fujii, Chem. Commun., 1997, 677–678 RSC; (d) L. A. Jenkins, J. K. Bashkin, J. D. Pennock, J. Florian and A. Warshel, Inorg. Chem., 1999, 38, 3215 CrossRef CAS; (e) K. P. McCue, D. A. Voss, C. Marks and J. R. Morrow, J. Chem. Soc., Dalton Trans., 1998, 2961 RSC; (f) C. Bazzicalupi, A. Bencini, E. Berni, A. Bianchi, V. Fedi, V. Fusi, C. Giorgi, P. Paoletti and B. Valtancoli, Inorg. Chem., 1999, 38, 4115 CrossRef CAS; (g) T. Beissel, K. S. Burger, G. Voigt, K. Wieghardt, C. Butzlaff and A. X. Trautwein, Inorg. Chem., 1993, 32, 124 CrossRef CAS; (h) J. N. Burstyn and K. A. Deal, Inorg. Chem., 1993, 32, 3585 CrossRef CAS; (i) W. H. Chapman and R. Breslow, J. Am. Chem. Soc., 1995, 117, 5462 CrossRef; (j) K. A. Deal and J. N. Burstyn, Inorg. Chem., 1996, 35, 2792 CrossRef CAS; (k) R. W. Hay and N. Govan, Polyhedron, 1998, 17, 463 CrossRef CAS; (l) E. L. Hegg, K. A. Deal, L. L. Kiessling and J. N. Burstyn, Inorg. Chem., 1997, 36, 1715 CrossRef CAS; (m) P. Jurek and A. E. Martell, Inorg. Chim. Acta, 1999, 287, 47 CrossRef CAS; (n) E. Kimura, H. Hashimoto and T. Koike, J. Am. Chem. Soc., 1996, 118, 10963 CrossRef CAS; (o) T. Koike, S. Kajitani, I. Nakamura, E. Kimura and M. Shiro, J. Am. Chem. Soc., 1995, 117, 1210 CrossRef CAS; (p) J. H. Suh, S. J. Son and M. P. Suh, Inorg. Chem., 1998, 37, 4872 CrossRef CAS; (q) C. Wendelstorf, S. Warzeska, E. Kovari and R. Kramer, J. Chem. Soc., Dalton Trans., 1996, 3087 RSC.
- (a) E. Baraniak, D. A. Buckingham, C. R. Clark, B. H. Moynihan and A. M. Sargeson, Inorg. Chem., 1986, 25, 3466 CrossRef CAS; (b) D. A. Buckingham and C. R. Clark, Inorg. Chem., 1986, 25, 3478 CrossRef CAS; (c) D. A. Buckingham, D. M. Foster and A. M. Sargeson, J. Am. Chem. Soc., 1969, 91, 3451 CrossRef CAS; (d) D. A. Buckingham, P. Morris, A. M. Sargeson and A. Zanella, Inorg. Chem., 1977, 16, 1910 CrossRef CAS; (e) J. H. Kim, J. Britten and J. Chin, J. Am. Chem. Soc,., 1993, 115, 3618 CrossRef CAS.
- (a) H. Aneetha, Y. H. Lai, S. C. Lin, K. Panneerselvam, T. H. Lu and C. S. Chung, J. Chem. Soc., Dalton Trans., 1999, 2885 RSC; (b) L. Tei, A. J. Blake, A. Bencini, B. Valtancoli, C. Wilson and M. Schroeder, Inorg. Chim. Acta, 2002, 337, 59 CrossRef.
- L. Siegfried, R. Kowallick and Th. A. Kaden, Supramol. Chem., 2001, 13, 357 CrossRef CAS.
- L. Tei, A. J. Blake, V. Lippolis, C. Wilson and M. Schroeder, Dalton Trans., 2003, 304 RSC.
- W. Schibler and Th. A. Kaden, J. Chem. Soc., Chem. Commun., 1981, 603 RSC.
- A. Comparone and Th. A. Kaden, Helv. Chim. Acta, 1998, 81, 1765 CrossRef CAS.
- G. Hänisch, Th. A. Kaden and A. D. Zuberbühler, Talanta, 1979, 26, 563 CrossRef.
- H. Gampp, M. Maeder, Ch. J. Meyer and A. D. Zuberbühler, Talanta, 1985, 32, 257 CAS.
- A. Altomare, G. Cascarano, G. Giacovazzo, A. Guagliardi, M. C. Burla, G. Polidori and M. Camalli, J. Appl. Crystallogr., .1994, 27, 435 Search PubMed.
- D. J. Watkin, C. K. Prout, R. J. Carruthers, P. Betteridge and R. I. Cooper, CRYSTALS, Issue 11, Chemical Crystallography Laboratory, Oxford, 2001 Search PubMed.
- L. J. Farrugia, J. Appl. Crystallogr., 1997, 30, 565 CrossRef CAS.
- B. Jung, Thesis; Universität Basel, 1995.
- Commercial Software.
- B. Bosnich, C. H. Poon and M. Tobe, Inorg. Chem., 1965, 4, 1102 CrossRef CAS.
- D. Tschudin, A. Basak and Th. A. Kaden, Helv. Chim. Acta, 1988, 71, 100 CAS.
| This journal is © The Royal Society of Chemistry 2005 |