Synthetic, self-assembly ABCD nanoparticles; a structural paradigm for viable synthetic non-viral vectors
Kostas
Kostarelos†
and
Andrew D.
Miller
Imperial College Genetic Therapies Centre, Department of Chemistry, Flowers Building, Armstrong Road, Imperial College London, London SW7 2AY, UK. E-mail: a.miller@imperial.ac.uk; Fax: +44 20 7594 5803
First published on 22nd September 2005
Abstract
Gene therapy research is still in trouble owing to a paucity of acceptable vector systems to deliver nucleic acids to patients for therapy. Viral vectors are efficient but may be too dangerous. Synthetic non-viral vectors are inherently safer but are currently not efficient enough to be clinically viable. The solution for gene therapy lies with improved synthetic non-viral vectors systems. This review is focused on synthetic cationic liposome/micelle-based non-viral vector systems and is a critical review written to illustrate the increasing importance of chemistry in gene therapy research. This review should be of primary interest to synthetic chemists and biomedical researchers keen to appreciate emerging technologies, but also to biological scientists who remain to be convinced about the relevance of chemistry to biology. (209 references.)
 Kostas Kostarelos | Dr Kostas Kostarelos obtained his Diploma and PhD from the Department of Chemical Engineering at Imperial College London (UK), in 1995. He was Assistant Professor of Genetic Medicine and Chemical Engineering in Medicine at Cornell University Weill Medical College (USA), then Deputy Director of the Imperial College Genetic Therapies Centre, London, UK. He is currently the Deputy Head of the Centre for Drug Delivery Research, The School of Pharmacy, University of London; and an Associate Member of the Imperial College Genetic Therapies Centre and the Tissue Engineering and Regenerative Medicine Centre, Chelsea & Westminster Hospital, Imperial College London. |
 Andrew David Miller | Prof. Andrew David Miller is both Professor of Organic Chemistry & Chemical Biology at Imperial College London (UK) and founding Director of the Imperial College Genetic Therapies Centre (GTC). He graduated from Bristol University in 1984 with a BSc degree, performed his PhD thesis research at the University of Cambridge (UK), and then carried out postdoctoral research at Harvard University (USA). Since 1990, Prof Miller has been a member of academic staff in the Chemistry Department of Imperial College London (UK) pioneering research into synthetic non-viral vector systems for gene therapy, the chemistry of stress and the proteomic code. In his career, he has received several awards and fellowships, and authored nearly 150 papers, reviews, book chapters and patent applications to date. Prof. Miller has also co-founded two GTC spin-out companies, Proteom Ltd in September 1999 and IC-Vec Ltd in December 2001. |
1. Introduction
Gene therapy may be described as the use of genes as medicines to treat disease, or more precisely as the delivery of nucleic acids by means of a vector to patients for some therapeutic purpose. Gene therapy is a therapeutic modality with enormous promise, but one that has to date failed regrettably to deliver much of therapeutic significance in spite of all the clinical interest.1 The primary reason for current failure and the ensuing frustration in the field is the inadequacy of vectors used to deliver therapeutic nucleic acids to their desired site of action in cells of the target organs of choice. Researchers have been seduced by the apparent simplicity of gene therapy approaches to treatment leading to a drive for clinical applications before vector technologies have been adequately developed or understood. Predictably, there has been a dramatic loss of confidence in gene therapy research in recent times matched by a decline in scientific and public perceptions of gene therapy. This is unhelpful, gene therapy retains all future promise but there now needs to be a period of patient, logical technical and scientific development of new vector systems prior to any major second round of clinical trial activity.1 This process is ongoing.1.1 Viral or non-viral?
Which type of new vector system should be most appropriate to develop, viral or non-viral, synthetic or physical? In our view, synthetic non-viral vector systems represent the only realistic choice for routine in vivo applications and gene therapy in the future. Synthetic non-viral vector systems have many potential advantages compared with viral systems, including significantly lower toxicity/immunogenicity and potential for oncogenicity, size independent delivery of nucleic acids (from oligonucleotides to artificial chromosomes), simpler quality control, and substantially easier pharmaceutical and regulatory requirements. Increasing public alarm particularly with viral vectors may also be strengthening these significant advantages. Ever present in the minds of the public and regulators is the potential for toxic side effects from the use of viral vectors. Therefore, basic clinical confidence in non-viral vectors is growing and the various advantages listed above inherent in synthetic non-viral vector systems should ensure substantial clinical uptake once the science and technology of these vector systems can be appropriately matured for routine clinical use. In our opinion, recent advances suggest that this process of maturation is now progressing with pace. Appropriate synthetic non-viral vector systems for in vivo applications and gene therapy should not now be far off in coming, and for reasons of lower toxicity if for nothing else, synthetic systems making use of cationic lipids (cytofectins) and liposomes should be paramount.1.2 Barriers to effective nucleic acid delivery
 | ||
| Fig. 1 Diagram to show process of LD particle cell entry. LD particles that have not succumbed to aggregation and/or serum-inactivation associate with the cell surface and enter usually by endocytosis. The majority in early endosomes become trapped in late endosomes (Path A) and the nucleic acids fail to reach the cytosol. A minority of are able to release their bound nucleic acids into the cytosol. Path B is followed by RNA that acts directly in the cytosol. Path C is followed by DNA that enters the nucleus in order to act. The diagram is drawn making the assumption that plasmid DNA has been delivered which is expressed in an epichromosomal manner. Reproduced with the permission of Bios Scientific Publications.21 | ||
These myriad problems ensure that LD particles, especially when delivered i.v., have very short circulation times (<minutes) in biological fluids. A direct consequence of short circulation times is the classic first pass effect. After i.v.-injection of LD complexes, gene expression in the lung is typically 100-fold greater then in other organs such as the liver or spleen.15 This is mainly due to the fact that the pulmonary circulation is the first capillary bed that LD complexes will encounter post-injection and enlarged serum-disrupted LD complexes will readily deposit in this lung microvasculature15–17 possibly anchored by association with heparin proteoglycans on the pulmonary endothelial surface.18 Cationic polymer–DNA (polyplex, PD) particles are also vulnerable in similar ways. Accordingly, any synthetic non-viral vector system that is intended to be viable for in vivo applications or gene therapy must at the very least be equipped with the capacity to evade these extracellular hazards and reach the desired cells in the target organ of choice.
These extracellular barriers discussed in this section of the review are far from exhaustive and are primarily valid as long as synthetic non-viral vector systems are involved in local delivery applications in vivo to lung, peritoneal cavity, vascular system or main filtration organs such as the liver. For systemic delivery to other organs including tumours, the circulatory barriers described above are just the beginning and significant issues concerning tissue penetration, cell organisation, and access to cells of interest through the extracellular matrix may become very significant.
LD particles are not cell-type specific, they may be slow to enter cells (hours), are prone to endosome entrapment, and appear only to be weak facilitors of DNA entry into the cell nucleus. Tseng et al.22 have provided convincing evidence that DNA entry within the nuclear envelope is impossible without the intervention of M-phase in the cell cycle when the nuclear membrane is partially dismantled to allow mitosis and cell division to take place. Nuclear-pore complexes appear unable to support facile entry of large pDNA into the nucleus. Indeed, the complexity of these pore complexes is only now being fully appreciated and access to the nuclear volume via these pore complexes should be regarded as one of the most severe barriers to effective DNA delivery to cells.23–27 A further barrier to efficient DNA delivery appears to be the vulnerability of exogeneous DNA to digestion by cytosolic nucleases once DNA has escaped into the cytosol from early endosome compartments.28,29 Studies involving fluorescence correlation spectroscopy have also revealed pDNA to bind extensively to immobile, cellular obstacles (cytoskeleton) in the cytosol thereby severely impeding intracellular migration of DNA towards the nucleus.30,31
Clearly, similar barriers are perceived for PD particles as well. Hence, any synthetic non-viral vector system that is intended to be viable for in vivo applications or gene therapy must at the very least be equipped with the capacity for rapid endosomal uptake followed by efficient endosmolysis, cytosolic trafficking and nuclear entry. Obviously, efficient nuclear entry is only required for DNA but not if RNA is involved. Conceivably, benefits could be had alternatively by avoiding endocytosis altogether and harnessing alternative cellular uptake mechanisms. However, this may well depend upon both vector characteristics and the nature of cells in the organs of choice that have been selected for nucleic acid delivery.
2. ABCD nanoparticles
In the light of the foregoing discussion and given the numerous permutations of synthetic non-viral vector systems that have been developed over the last few years, there is a need to find a common language with which to discuss and appreciate these systems in a framework that allows us to relate different, individual systems and hence derive meaningful and realistic structure–activity correlations. Therefore, we would like to introduce the self-asscmbly ABCD nanoparticle concept as an appropriate structural paradigm for synthetic non-viral vector systems used for in vitro, ex vivo and/or in vivo applications (Fig. 2). In ABCD nanoparticles, nucleic acids (A) are condensed within functional concentric layers of chemical components designed for biological targeting (D), biological stability (C) and cellular entry/intracellular trafficking (B). For the purposes of this review, the AB core particle comprises nucleic acids (either DNA or RNA) (A) condensed and/or encapsulated by liposomes/micelles (B) in a non-covalent manner. Typically DNA may be in the form of pDNA or oligodeoxynucleotide (ODN) and RNA could be in the form of messenger RNA (mRNA), oligonucleotide (ON) or small interference RNA (siRNA) for instance. AB core particles in our new nomenclature can be equivalent to LD (or even PD) particles and so should be expected to be appropriate for functional delivery of nucleic acids in vitro, perhaps ex vivo with limited applications in vivo. | ||
| Fig. 2 ABCD nanoparticle concept. Graphic illustration of ABCD nanoparticle structure to show how nucleic acids (A) are condensed in functional concentric layers of chemical components purpose designed for biological targeting (D), biological stability (C), and cellular entry (B). Reproduced with the kind permission of Elsevier Academic Press.209 | ||
For in vivo use, a stealth/biocompatibility polymer (C-layer) should be required that needs to be introduced by attachment to the surface of each AB core particle thereby conferring colloidal and structural integrity to AB core particles in biological fluids. Finally, biological-targeting ligands may be required as part of an optional exterior coating (D-layer) designed for the active targeting of nanoparticles firstly to the organ of choice in vivo but preferably to target cells of interest within the organ of choice. The requirement for biological-targeting ligands may not be obligatory. There are viable ABC nanoparticle systems that enter organs of choice in vivo by a process of passive targeting, namely organ/tissue accumulation through biophysical means without the need for active biological-targeting ligands. However, active biological-targeting processes are expected to subvert passive-targeting processes and “reprogram” nanoparticle systems to accumulate in alternative organs of choice with more precision, speed and efficiency than passive targeting processes will allow.
3. AB core particles
3.1 Cytofectins
AB core particles are equivalent to LD particles or other such particles generated by the condensation and/or encapsulation of nucleic acids. By far the majority of AB core particles have been generated by the combination of simple cationic liposome/micelle systems with pDNA. Simple cationic liposome/micelle systems are formed from either a single synthetic cationic amphiphile (known as a cytofectin; cyto- for cell and -fectin for transfection [i.e., gene delivery and expression]) or more commonly from the combination of a cytofectin and a neutral lipid such as dioleoyl L-α-phosphatidylethanolamine (DOPE) 1 or cholesterol (Chol) 2 (Fig. 3). There are impressive numbers of cytofectins already described in the literature and available commercially19,20,32–34 but all have in common a hydrophobic moiety covalently attached to a hydrophilic moiety through a polar linker (Fig. 3). Whilst hydrophobic regions are reasonably similar, polar linkers and cationic head groups vary quite substantially. The structures of a number of cytofectins are shown illustrating the structural diversity that is tolerated without necessarily impairing the efficiency of transfection (Fig. 3)! The cytofectin field now appears to be approaching saturation so that the creation of further novel structures is now much less likely to make a novel contribution unless their preparation is associated with the onward generation of ABC and ABCD nanoparticle systems for in vivo applications. | ||
| Fig. 3 Cytofectins and neutral lipids. Summary of important cytofectins and neutral lipids that are mentioned in the text. | ||
Nevertheless, there are a small number of recent additions to the cytofectin-pantheon that are worth mentioning. N1-cholesteryloxycarbonyl-3,7-diazanonane-1,9-diamine (CDAN) 9 (Fig. 3) syntheses and properties have been described a number of times.35–37 However, unusually CDAN/DOPE cationic liposomes (1 ∶ 1 m/m, Trojene™) have now been found to mediate very high efficiency pDNA delivery to cells in vitro in the presence of cell growth medium, hence allowing for mininum-handling transfection protocols to be developed.38 Furthermore, CDAN/DOPE cationic liposomes (45 ∶ 55 m/m, siFECTamine®) have also been shown to facilitate the delivery of siRNA in vitro to cells with even more effect, as shall also be described later in more detail.39 For this reason, an attractive new solid phase methodology was very recently devised for the synthesis of CDAN in excellent yield that represents an effective synthesis of this increasingly critical cytofectin40 (Scheme 1). Balaban and coworkers,41,42 have reported functional solution-phase syntheses of novel pyridinium amphiphiles that enlarge on the contributions of others in this important area of cytofectin design,43,44 that is also growing in importance (Scheme 2). Multipurpose Gemini surfactants have also found their way into cytofectins45,46 (Scheme 3). Finally, one of the most innovative new cytofectins to emerge has been from Thompson and coworkers. They developed O-(2R-1,2-di-O-(1′Z,9′Z-octadecadienyl)-glycerol)-N-(bis-2-aminoethyl)-carbamate (BCAT) based on plasmenylcholine synthesis, whose enol ether linkages are primed for acid-catalysed hydrolysis in conditions of acid pH47–49 (Scheme 4). The notional design objective was to ensure that BCAT should mediate DNA delivery to cells. Thereafter, acid-catalysed BCAT decomposition was expected to take place in acidic endosome compartments leading to enhanced endosmolysis thereby increasing the proportion of bound nucleic acids able to escape from early-endosomes into the cytosol. This approach to the endosomal barrier problem (outlined above) appears to have been less effective than expected owing to the unexpectedly slow rate of enol-ether hydrolysis at pH 5.5. This is unfortunate, but Thompson and coworkers are already at work innovating the next generation of acid-sensitive functional groups.
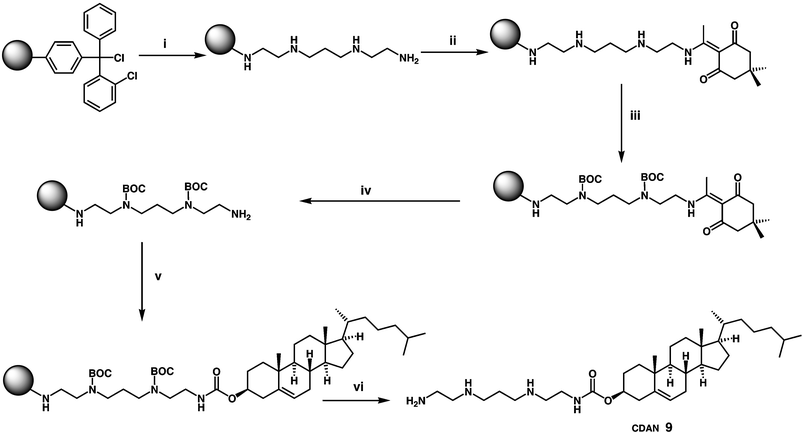 | ||
| Scheme 1 Reagents and conditions: (i) polyamine (10 equiv.), CH2Cl2, rt, 2h, then MeOH (1000 equiv.), rt, 10 min; (ii) Dde-OH (10 equiv.), DMF, rt, 12 h; (iii) Boc2O (5 equiv. per free amine), NEt3 (2 equiv. per free amine), CH2Cl2, rt, 4 h; (iv) 2% hydrazine hydrate in DMF, rt, 10 min (rpt step × 2); (v) cholesterol chloroformate (10 equiv.), NEt3 (3 equiv.), rt, 4 h; (vi) 50% TFA in CH2Cl2, rt, 1 h, 93%.40 | ||
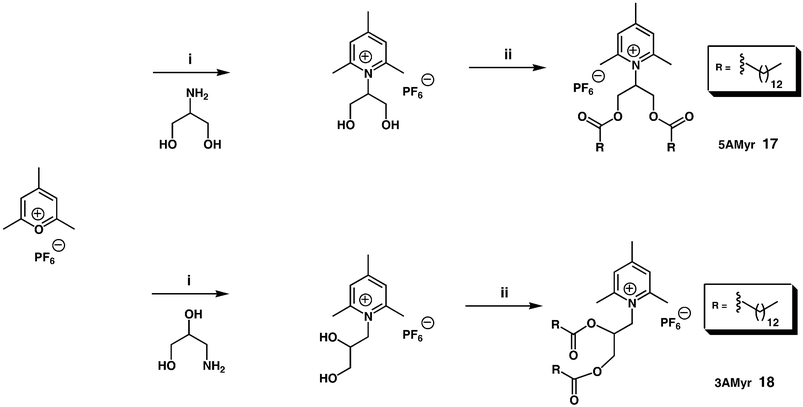 | ||
| Scheme 2 Reagents and conditions: (i) NEt3 (1.2 equiv.), EtOH/AcOH, 1–3 h, 50–85%; (ii) NEt3 (2 equiv.), myristoyl-Cl (2.2 equiv.), AcCN, 3–5h, reflux.42 | ||
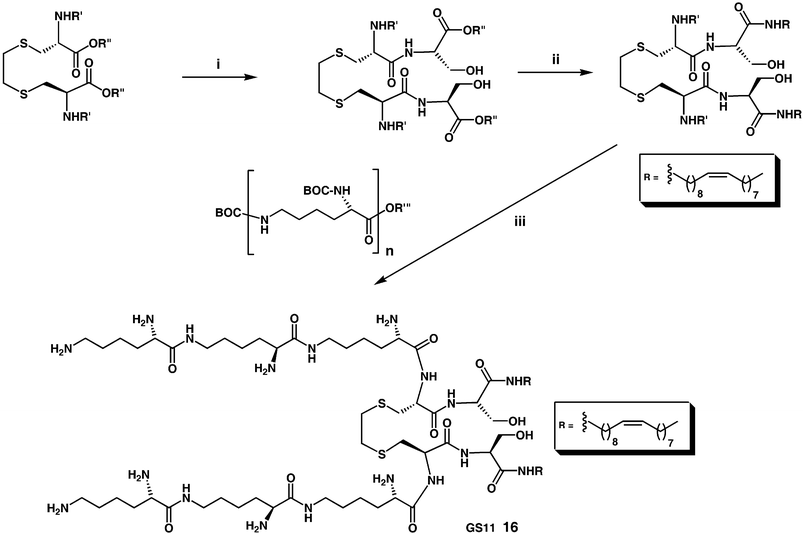 | ||
| Scheme 3 Reagents and conditions: (i) L-serine (2 equiv.), K2CO3 (2 equiv.), THF/H2O 1 ∶ 1 v/v, rt, 72 h; (ii) (a) NHS (2 equiv.), DCC (2 equiv.), THF, rt, 24 h (b) NEt3 (2 equiv.), oleylamine (2 equiv.), THF, rt, 48h; (iii) (a) BOC-protected L-K3 (R‴ = N-succinimido) (>2 equiv.), H2O/NaOH/THF, rt, 48 h (b) MeOH/HCl.45 | ||
 | ||
| Scheme 4 Reagents and conditions: (i) TBDPS-Cl, imidazole; (ii) HCl, MeOH; (iii) oleoyl-Cl, pyridine; (iv) LDA; (v) Et2POCl, HMPA; (vi) Pd(PPh3)4, Et3Al; (vii) TBAF, TBAH; (viii) (a) py2CO, NEt3 (b) N, N′-dipthalamidylethylenetriamine; (ix) N2H4–H2O.49 | ||
3.2 Characteristics of LD particles
Typically, cytofectin and neutral lipid components are mixed together in an appropriate mol ratio and then induced or formulated into unilammellar vesicles by any one of a number of methods including reverse phase evaporation (REV), dehydration–rehydration (DRV) and extrusion.19,20,37 Alternatively, cytofectins may be assembled into micellar structures after being dispersed in water or aqueous organic solvents.19,20 Unilamellar vesicles or micelles may then be combined with nucleic acids to form nanometric LD particles. Biophysical structure–activity studies designed to understand the structures of LD particles and their relationships to LD transfection efficiency have been numerous. Unfortunately, the diversity of cytofectin structures, LD particles and biological targets has resulted in considerable inconsistency in the results reported by the research groups concerned. For instance, LD mixtures with a positive/negative charge ratio higher than 1,50,51 or close to 152–55 have been reported to be optimal for LD transfection in vitro. In direct contrast, LD mixtures with an overall positive/negative charge ratio of <1 appear to be optimal for LD transfections of COS-7 cells in vitro and even of Balb/c mice lungs in vivo.36 These latter observations have been supported by the results of others too.56Similarly diverse views exist concerning the structures of LD mixtures optimal for transfection. In some circumstances, LD mixtures optimal for in vitro transfection appear to be heterogeneous and consist of a variety of particles and other structures all in dynamic equilibrium.57,58 These particles and other structures have been variously identified and described by a number of researchers and they include multilamellar lipid/nucleic acid clusters (>100 nm in diameter)59–63 perhaps with some surface associated nucleic acids,64 or with thinly lipid-coated DNA nucleic acid strands65 and even in the presence of free nucleic acids.61 Such structural observations have led to a substantive debate concerning the relative importance of each of these structural entities for efficient in vitro transfection.
However, LD mixtures optimal for in vitro transfection do not necessarily have to be heterogeneous and substantially polydisperse. Our recent studies using cryo-electron microscopy have clearly demonstrated that LD mixtures optimal for in vitro and in vivo transfection may actually consist of discrete LD particles (size range; 60–250 nm in diameter) exhibiting bilamellar perimeters and striations with a periodicity of 4.2 ± 2 nm (Fig. 4).36 Small-angle X-ray scattering (SAXS) and other cryo-electron microscopy studies of LD mixtures have revealed similar periodicities of approx. 6.5 and 3.5 nm that have been shown to result from the encapsulation of DNA molecules in regular periodic arrays within a multilamellar LD assembly59,60,66–68 (Fig. 5). Therefore, the observed LD particles are most likely composed in a similar way. Hence in this case at least, optimal LD transfection in vitro and in vivo must be primarily mediated by these discrete, multilammellar LD particles.36 The significance of discrete LD particles for optimal transfection has been supported by the results of at least one other published study comparing LD structure with in vivo transfection efficacy.69 Evidence then suggests that the regular multilamellar bilayer structure (LαI) of LD particles should undergo a phase change in the endosome forming inverted hexagonal phase structures (HII) that may disrupt endosome membranes and faciliate nucleic acid escape into the cytosol70 (Fig. 5). Lipids like DOPE are well known to prefer HII phases under physiological conditions of temperature and pH, and the LαI → HII phase transition has been widely implicated as a key facilitator of membrane fusion and membrane disruption events. Hence the inclusion of DOPE in an LD system is likely to facilitate endosome escape of bound nucleic acids through induced LαI → HII phase destabilisation.71
![Cryo-electron microscopy images of LD particles. These LD particles were formed after the combination of CDAN/DOPE cationic liposomes and pDNA in the [cytofectin]/[nucleotide]
([cyt]/[nt]) mol ratio of 0.6, optimal for in vitro and in vivo lung transfection. Final lipid concentration was 0.17 mM. Magnification is 150 000×
(1 cm = 67 nm).36](/image/article/2005/CS/b307062j/b307062j-f4.gif) | ||
Fig. 4 Cryo-electron microscopy images of LD particles. These LD particles were formed after the combination of CDAN/DOPE cationic liposomes and pDNA in the [cytofectin]/[nucleotide]
([cyt]/[nt]) mol ratio of 0.6, optimal for in vitro and in vivo lung transfection. Final lipid concentration was 0.17 mM. Magnification is 150![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000×
(1 cm = 67 nm).36 000×
(1 cm = 67 nm).36 | ||
 | ||
| Fig. 5 LD particle internal structure and dynamics. Left-hand side: schematic of the lamellar LαI phase of DNA molecules interacting with cationic bilayers forming a multilayered assembly typical of LD particle composition. DNA double helices are shown as ribbons (blue and purple), head groups of anionic/zwitterionic lipids are shown as white spheres while those of cytofectins are shown as grey spheres. The notation δm refers to bilayer thickness, δw to interbilayer separation and dDNA to DNA interaxial spacing. Right-hand side: conversion from lamellar LαI phase to the columnar, inverted hexagonal HII phase thought to be typical of LD particle composition during the transfection process, takes place by two possible routes. The first involves pathway (I) typified by negative curvature Co induced in each cationic monolayer due to the presence of DOPE 1. The second involves pathway (II) typified by loss in membrane rigidity κ thereby encouraging phase inversion. Reproduced with permission from the American Association for the Advancement of Science.70 | ||
General biophysical structure–activity studies have generated few proper correlations between LD particles structure, physical attributes and transfection efficiency in vitro,33 and in general there have been few attempts to derive unifying biophysical parameters able to account for differences in LD transfection efficiency in vitro, yet alone relate these parameters to in vivo transfection performance. One exception may be found in the work of Stewart et al.36 wherein the physical properties of a systematic series of cationic liposomes and their corresponding LD mixtures were studied. Liposomes were formulated from DOPE 1 and cholesterol-based polyamine cytofectins such as CDAN 9 (Fig. 3). Successful in vitro transfection was linked to the ability of cationic liposome systems to (1) provide relatively inefficient neutralisation, condensation and encapsulation of nucleic acids into LD particles; and (2) present unprotonated amine functional groups (pKa < 8) at neutral pH with the capacity for substantial endosome buffering, thereby enabling the osmotic shock mechanism to facilitate nucleic acid escape from endosome compartments as their internal pH is reduced from pH 7 to 5.5.72,73 Critically, both main factors were observed to be under the control of the cytofectin polyamine head group structure. The inclusion of “natural” propylene and butylene spacings between the amine functional groups of head groups appeared to promote efficient neutralisation, condensation and encapsulation of nucleic acid. Inclusion of “unnatural” ethylene spacings appeared to promote the reverse effect although at the same time assisting the perturbation of amine pKa values from 9–10 to below 7. The appearance of such perturbed pKa values now appears not only to be important to enable the osmotic shock mechanism for endosome escape but also to render LD particles metastable and prone to partial aggregation and sedimentation onto cell surfaces in vitro.38 Such sedimentation is likely to be beneficial for transfection unless the given LD particles themselves induce cytotoxicity. Consistent with this observation, a correlation has also been established between transfection efficiency and enhanced membrane fluidity in both lipoplex and cellular membranes.74
3.3 Ternary LD particles
Ternary LD particles represent another form of AB nanoparticle system. In these systems the cationic character of the cationic liposome/micelle system used to condense and/or encapsulate nucleic acids is supplemented by an additional cationic entity. In the case of pDNA, this cationic entity is frequently used to precondense the pDNA prior to final condensation and encapsulation by cationic liposomes/micelles. This is particularly true of the lipid:protamine:DNA (LPD) system prepared using the salmon sperm-derived peptide protamine,5,75–83 and the liposome:mu:DNA (LMD) system prepared from adenoviral derived peptide μ (mu).37 Other cationic entities that have been used to condense nucleic acids prior to complexation with cationic liposomes/micelles include poly-L-lysine (pLL),84,85 spermidine,86 lipopolylysine,87 histone proteins,88 chromatin proteins,89 human histone derived peptides,90L-lysine containing synthetic peptides,91 not to mention a histidine/lysine (H-K) copolymer.92 The formulation process of LMD is illustrated (Fig. 6). LPD particles may be prepared in a similar way. | ||
| Fig. 6 LMD formulation. Schematic illustration of LMD particle formation. Initially, pDNA (D) is introduced under vortex mixing to mu peptide (M) in the ratio of 0.6∶1 w/w forming MD particles. These themselves are then added under vortex mixing to cationic liposomes (L) in a ratio of 12∶1 w/w with respect to pDNA, resulting in the formation of bilamellar LMD particles. Inset: cryo-electron microscopy image of LMD particle prepared with DC-Chol/DOPE cationic liposomes and pDNA (1 cm = 60 nm). Reproduced with the kind permission of Elsevier Academic Press.37 | ||
Mature adenovirus consists of an icosahedral, non-enveloped capsid particle (approx. 90 nm) enclosing a core complex that consists of a linear dsDNA viral genome (∼36 kbp) non-covalently associated with two cationic proteins (proteins V [pV] and VII [pVII]) and the 19-residue mu peptide.93,94 Mechanistic studies using the mu peptide have revealed how increasing pDNA–peptide interactions lead to progressive base-pair-tilting generating regions of high and low double helical stability, that in turn promote super-coiling followed by pDNA hydrophobic collapse.95,96 In kinetic terms, the process of pDNA condensation and the reverse process of pDNA expansion appear to be equivalent to small single domain protein folding and unfolding respectively.96 Chaotic behavior is also observed at low peptide/pDNA ratios (0.1–0.3 w/w) that becomes more uniform at higher ratios suggesting that with suboptimal ratios, pDNA is condensing in a multitude of conformations, each representing different stages of hydrophobic collapse in the search for the thermodynamically most stable (i.e., the fully condensed pDNA molecule). This represents yet another analogy with protein folding. At higher ratios, peptide/pDNA complexes formed appear to be increasingly irreversible consistent with the formation of kinetically and/or thermodynamically stable, condensed pDNA molecules.96 Such stable states could create problems for the successful transcription of DNA post delivery to cells, yet another barrier to successful delivery of DNA to cells that is yet to be understood!
Both LPD and LMD systems are able to form discrete, essentially mono-disperse (single-size) particles. DOTAP/Chol-based LPD systems were even more effective and were found to formulate into discrete, essentially single-size particles (approx 135 ± 42 nm).77 DC-Chol/DOPE-based LMD systems were found to formulate into discrete single-size particles (approx. 120 ± 30 nm)37 (Fig. 6). LMD particles can be formulated reproducibly that are amenable to long-term storage at −80 °C and stable up to a pDNA concentration of 5 mg ml−1 (nucleotide concentration 15 mM), a concentration appropriate for facile use in vivo.37 Using LD systems, nucleotide concentrations >4 mM are difficult to achieve owing to ready LD particle aggregation above this concentration threshold.35,36,97 Moreover, LMD transfections appear to be significantly more time and dose efficient in vitro than LD transfections. LMD transfection times as short as 10 min and DNA doses as low as 0.001 µg per well result in significant gene expression. Furthermore, LMD transfections will also take place in the presence of biological fluids (e.g., up to 100% serum), conditions typically intractable to LD transfections, suggesting that LMD formulations exhibit an additional element of stability. In consequence, LMD transfection of murine lung in vivo was up to six-fold more dose efficient than transfection with GL-67/DOPE/DMPE-PEG5000 (1 ∶ 2 ∶ 0.05 m/m/m) (one of the best synthetic non-viral vector systems reported to date for lung transfection). LMD has been called an artificial virus-like nanoparticle (VNP) on the basis that cryo-electron microscopy shows LMD particles to consist of a mu:DNA (MD) particle encapsulated within a cationic bilamellar liposome (Fig. 6).
However, this additional element of LMD stability is unlikely to be adequate for general in vivo applications and gene therapy.98 Indeed, corresponding LPD particles are readily modified by serum causing gradual vector disintegration, release of DNA and probable RES scavenging.5,77 Released DNA is also noted to be susceptible to extracellular nuclease digestion. Furthermore, LPD particles have been found to promote a systemic, Th1-like innate immune response in mice, much more appropriate for a DNA vaccine than for gene therapy.79 However, the general impression given is that LPD like LMD systems could have a role to play clinically for the passive delivery of genes to lung but are not appropriate for targeted gene delivery to other tissues.5
Studies carried out by confocal microscopy on dividing tracheal epithelial cells suggest that endocytosis is not a significant barrier to LMD transfection. However, the nuclear envelope remains a highly significant barrier. LMD particles were found to enter cells rapidly (minutes), and disintegrate almost immediately leaving mu peptide free to migrate to the nuclear zone (within 15 min) and pDNA to enter after a further 15–30 min. There is every possibility that both cytofectin and perhaps even mu peptide are exercising fusogenic behaviour with respect to early endosome membranes.99–101 However, LMD does not appear to facilitate pDNA entry into the nucleus of growth arrested (aphidicolin-treated) cells suggesting that the nuclear pore complex remains a significant barrier to LMD transfection even though mu peptide has been shown to possess strong nuclear localisation sequence (NLS) characteristics.102 The obvious solution is to ensure that mu peptide and pDNA remain in association for long enough within non-dividing (quiescent) cells for the DNA to utilise the NLS characteristics of the mu peptide to cross the nuclear membrane.95,102 Evidence from DNA trafficking and expression studies using NLS peptides covalently or non-covalently associated with the pDNA appear to support this suggestion amply,27,103 assisted by the presence of such elements as the SV40 enhancer in pDNA structure.104
Very recently, a new ternary LD system known as the multifunctional envelope-type nano-device (MEND) system was described.105 The formulation process compares in an interesting way to the LMD and LPD systems involving a cationic DNA/polycation complex interacting with an anionic fusogenic lipid film prior to sonication into large but discrete particles (402 ± 73 nm) whose charge can be modified by the post-insertion of stearyl octa-arginine (STR-R8) to give transfection competent particles (Fig. 7). Without doubt an imaginative, alternative way to arrive at condensed discrete particles. In the cases of LMD, LPD and perhaps MEND particles, these represent systems that can be formulated in a reproducible and scalable manner, that are resistant to aggregation in low ionic strength media, are amenable to long term storage and give properly reproducible transfection outcomes. Therefore, these are ideal platforms upon which to build viable lipid-based synthetic, non-viral vector systems for DNA delivery in vivo by a process of modular upgrading through systematic chemical adaptation with appropriate tool-kits of known or newly designed chemical components.
 | ||
| Fig. 7 MEND formulation. Schematic illustration of MEND particle formation. Initially, cationic PD particles are formed from pDNA (D) and cationic polymer (P) (usually pLL). These associate electrostatically with a negatively charged mono-layer lipid film and are then encouraged to form particles by a process of hydration and sonication. Final post-modification with STR-R8 results in the formulation of cationic MEND particles (see text for references). Reproduced with the kind permission of Elsevier Academic Press.209 | ||
4 ABD particles
4.1 Synthetic ABD particles
Some fascinating examples of ABD particles have emerged in recent years notable for some in vivo viability although somewhat irregular in formulation. For instance, peptides consisting of an oligo-L-lysine moiety linked to a peptide moiety specific for cell surface integrin proteins have been combined with LD systems.106–109 In the latter case, credible enhancements of at least an order of magnitude in in vitro transfection have been observed over and above the results of binary LD transfection owing to the involvement of integrin-mediated cell uptake.107–109 Furthermore, enhancements to in vivo transfection have been reported as well, but the mechanism of these so-called lipid:integrin-targeting peptide:DNA (LID) systems does not actually appear to be integrin-receptor dependent in this case.106Modular adaptation of LMD particles has arguably resulted in alternative ABD systems whose behaviour has given more clarity. A glyco-LMD variant was prepared by a post-modification strategy in which neoglycolipid micelles were combined with pre-formulated LMD particles (AB system) in order to encourage insertion of neoglycolipid molecules into the outer leaflet membranes of LMD particles using their hydrophobic lipid moieties. The syntheses of neoglycolipids is shown given the particular use of an aminoxy functional group to couple reducing sugars to the lipid moiety without the requirement for any protecting groups illustrating the high chemoselectivity of the coupling reaction (Scheme 5). This coupling reaction takes place in aqueous as well as organic solvents, ideal given the range of reducing sugars coupled.98 The resulting glyo-LMD particles (ABD nanoparticles) were stable in high-salt medium (but not 100% serum) and mediated enhanced non-specific transfection of cells in vitro.98
 | ||
| Scheme 5 Reagents and conditions: (i) CH2Cl2, HO(CH2)2NH2 (2.2 equiv.), 10 h, 97%; (ii) (a) CH2Cl2, 0 °C, NEt3 (3 equiv.), MsCl (2.5 equiv.), 10 min; then 1 h at rt, 98% (b) THF, HO(CH2)3NH2 (10 equiv.), 6 h, 96% (c) CH2Cl2, NEt3, Boc2O, rt, 5 h, 90%; (iii) (a) CH2Cl2, 0 °C, NEt3 (3 equiv.), MsCl (2.5 equiv.), 10 min; then 2 h at rt, 90% (b) DMF, 80 °C, NaN3 (5 equiv.), NaI (1 equiv.), 3 h, 95% (c) THF, PMe3 (1.15 equiv.), rt, 3h (d) H2O/NH3, 88%; (iv) CH2Cl2, NEt3, Boc2O, rt, 5 h, 98%; (v) (a) EtOAc, NHS (1 equiv.), DCC (1 equiv.), 10 h, rt (b), EtOAc/THF 95/5 v/v, NEt3 (pH8), 2 h, rt, 90%; (vi) CH2Cl2, TFA (15 equiv.), 0 °C, N2, 5 h, 86%; (vii) saccharide, AcOH/DMF 1/1 v/v, rt.98 | ||
A peptido-LMD variant was also prepared very recently by a pre-modification strategy in which lipopeptides of two classes were formulated into cationic liposomes prior to LMD formulation. LMD formulations were prepared using both CDAN 9 and DC-Chol 8 cytofectins, The synthesis of one lipopeptide is shown, notable for the application of a novel variation of the Fukuyama–Mitsunobu reaction (Scheme 6), that now appears to have general applications in the synthesis of complex secondary amines.110 The peptide sequence used (tenascin peptide sequence: PLAEIDGIELA) was previously shown to target α9β1-integrin proteins predominant on upper airway epithelial cells in mammals. When peptido-LMD systems were prepared using CDAN 9 cytofectin, no evidence of receptor-mediated enhancement of transfection was observed. Instead, even with as little as 0.05 mol% of lipopeptide present in each peptido-LMD particle, transfection was at least 10-fold more effective than found for corresponding LMD systems without peptide present, irrespective of whether the cells under investigation expressed α9β1-integrin proteins or not! Such non-specific peptide enhancement may be interesting but is not necessarily desirable. When peptido-LMD systems were prepared using DC-Chol 8 cytofectin, thereby reducing the overall positive charge of each particle, a modest element of specific enhancement was observed (2-fold) over a general background enhancement that was otherwise non-specific.110
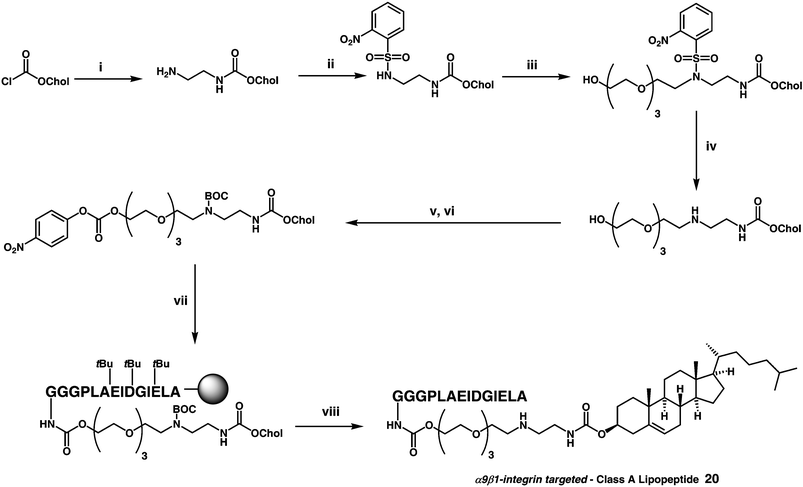 | ||
| Scheme 6 Reagents and conditions: (i) H2NCH2CH2NH2 (200 equiv.), 2 days, 65%; (ii) (a) 2-NsCl (1.3 equiv.), NEt3 (1.5 equiv.), CH2Cl2, 14 h, 87% (b) BnBr (1.1 equiv.), Ag2O (1.5 equiv.), 20 h, generating 55%; (iii) tetraethyleneglycol (TEG) (1.3 equiv.), DTBAD (1.5 equiv.) slow addition over 1 h in CH2Cl2, PPh2py (1.5 equiv.), CH2Cl2, 3 h, 71%; (iv) Na (10 equiv.), C10H8 (10 equiv.), −30 °C, 45 min, 74%; (v) Boc2O (1 equiv.), NEt3 (1.1 equiv.), CH2Cl2, 10 h, 84%; (vi) NEt3 (2 equiv.), DMAP (2 equiv.), p-nitrophenyl chloroformate (3 equiv.), CH2Cl2, 10 h, 92%; (vii) fully protected peptide (Fmoc deprotected on N-terminus) (0.5 equiv.), NEt3 (2.5 equiv.), DMF, 18 h; (viii) 95% v/v TFA/H2O, 90 min, 10% over two steps.110 | ||
Non-specific enhancements appear to be a hazard (or perhaps even an advantage under some circumstances) of using ABD systems. There has been some apparent success in using neoglycolipids as D-layer targeting agents inserted into LD complexes. For instance, Behr and coworkers reported in vitro galactose-receptor mediated uptake into hepatoma cells of LD complexes formulated with a triantennary galactolipid.50 By contrast, Kawakami et al. have suggested liver targeting in vivo using LD complexes formulated with a mannosyl neoglycolipid as targeting agent. However, non-specific mannosyl induced LD stabilisation leading to longer circulation times seems to be a sufficient explanation to account for these results too.111,112 An additional example of non-specific enhancements in ABD systems has been provided from experiments with the Transferrin (Tf) protein. Tf has been used quite frequently for D-layer biological targeting on the basis that transferrin receptors (TfR) are found routinely at the surface of vascular endothelial cells associated with tumours or the blood brain barrier and are rapidly internalised upon binding to Tf.113–118 However, whilst in vitro and ex vivo transfections with a Tf-LD ABD system are enhanced relative to binary LD transfection, the mechanism is quite clearly TfR independent, the protein instead acting primarily to promote endosome disruption and subsequent escape of complexed DNA into the cytosol,113,114 and even entry into the nucleus.119 In addition, Tf is an acidic protein, negatively charged at neutral pH. Accordingly, the association of Tf with binary LD systems seems to reduce the overall positive charge and provides simultaneously a combined stereo-electronic barrier to biological fluid components allowing in vitro transfection to take place, even in 60% serum. In the latter context, human serum albumin (HSA) has been deliberately combined with binary LD systems in order to create negatively charged, sterically protected complexes appropriate for in vitro transfection in the presence of up to 30% serum and even for lung or spleen transfection in vivo.120 Lectin proteins also promote in vitro transfection in the same context.121
Chang and coworkers have suggested that a simple reformulation of Tf-LD ABD systems is sufficient to give highly compact particles with a relatively uniform size (50–90 nm) comprising a dense Tf-DNA core enveloped by a membrane coated with Tf molecules spiking the surface. This system appears to render enhanced stability, improved in vivo gene transfer efficiency, and long-term efficacy for systemic p53 gene therapy of human prostate cancer when used in combination with conventional radiotherapy.122 Others have reported the need to introduce protamine giving the equivalent of a Tf-LPD ABD system that is able to transfect cells in a number of tissues post i.v. injection,123 otherwise alternative Tf-LD ABD systems have been administered directly (intra-tumoural injection) to subcutaneous mouse xenograft models of human prostate cancer,124 or else by i.a. administration into hepatocellular carcinoma.125 Such data is certainly consistent with the possibility of some TfR-mediated uptake of Tf-LD particles by some tissues.126
Chang and coworkers have also developed alternatives to the Tf-LD ABD systems. For instance, using an anti-TfR antibody variable region fragment (anti-TfR scFv), they have produced an anti-TfR scFv-LD system with the scFv covalently attached to a number of cytofectins that appears to show some promise for systemic p53 gene therapy in a number of human tumour models including human breast cancer metastasis.127,128 This anti-TfR scFv-LD system has been further improved by the inclusion of a cationic peptide (HoKC) to precondense pDNA during complex formation (i.e., in a similar way to LMD and LPD systems).129 However, the final coup-de-grâce has been to demonstrate that anti-TfR scFv-containing ABD systems are in fact inferior to a complete ABCD system in which a post-coating strategy was employed taking pre-formed LD particles (AB core particles) that were sequentially conjugated with PEG polymer (C-layer) and then anti-TfR scFv (D-layer).130 However, there may yet be a future for simple monoclonal antibody (MAb)-LD ABD systems with or without covalent coupling of the antibody to cytofectins,131,132 and even for ABD systems with associated lectins.121
4.2 Semi-synthetic ABD particles
Difficulties experienced in working with fully synthetic ABD particles have also resulted in the development of some semi-synthetic virosome systems. The term virosome was originally coined in reference to combinations of liposomes and various virus glycoproteins but is now more generally used to refer to various types of viral/non-viral hybrid vector systems. Of these the HVJ-liposome system is instructive. This semi-synthetic system is prepared from a combination of UV-irradiated virions of the Hemagglutinating Virus of Japan (HVJ; Sendai virus) and liposomes in which are encapsulated nucleic acids complexed with the High Mobility Group 1 (HMG-1) protein.133,134 The HMG-1 protein is there to assist nuclear access and localisation of delivered nucleic acids as well as promoting gene stabilisation within the nuclear envelope.133,135 Conventional HVJ-liposomes are negatively charged,133,134,136,137 however an HVJ-cationic liposome system has recently been developed, based on the cytofectin DC-Chol 8, that appears to transfect various mammalian cell types in vitro 100–800 fold more effectively than conventional HVJ liposomes.138 In addition, HVJ-cationic liposomes prepared with the cytofectin N-(α-trimethylammonioacetyl)-didodecyl-D-glutamate chloride (TMAG) 14 (Fig. 3), have also proved able to mediate delivery of nucleic acids to tracheal and bronchiolar epithelial cells in vivo with reasonable efficiency.139 One major reason for the success of HVJ-liposome systems is the presence of the hemagglutinin-neuraminidase (HN) and fusion (F) glycoproteins in the liposome bilayer (Fig. 8). These are fusogenic proteins that allow HVJ-liposomes to interact with cell surface sialic residues, fuse with the cell membrane and then release encapsulated nucleic acids directly into the cytosol, bypassing endocytosis altogether.134 For this reason, HVJ liposomes have also been called fusogenic liposomes.140 The clear success of HVJ-cationic liposomes has resulted in the development of a number of other cationic virosome systems including systems prepared with the influenza membrane fusion protein hemagglutinin that were used to deliver genes to cells in vitro,141 cationic lipid-reconstituted influenza-virus envelopes used to deliver an ODN to cells in vitro,142 and LD complexes prepared from DOTMA/DOPE cationic liposomes and pDNA doped with the partially purified G glycoprotein of the vesicular stomatitis virus envelope (VsV-G).143 These semi-synthetic ABD nanoparticle systems are sure to remain of great interest going forward with one caveat. Virosomes contain virus proteins that could be very immunogenic in vivo, thereby creating potentially serious problems for repeated use of virosome vectors as for complete viral vectors. | ||
| Fig. 8 HVJ-cationic liposome system. Cytofectins are incorporated in the lipid envelope. Reproduced with the kind permission of Bios Scientific Publishers.21 | ||
5 ABC particles
Particles of this type appear to be altogether more promising for in vivo applications and gene therapy owing to passive targeting of particles stabilised with the aid of C-layer stealth/biocompatibility polymers. Passive targeting is the process by which stabilised nanoparticles accumulate with time into organs possessing enhanced microvasculature (such as solid tumours, infection, and inflammation sites) by means of long-term circulation in biological fluids, without the requirement for D-layer active targeting agents. By far the most popular C-layer molecule is polyethyleneglycol (PEG). PEG provides a steric barrier to interaction with biological fluid components and prevents uptake of liposomal vesicles by cells of the RES.19,144 Safinya and coworkers have recently demonstrated that only PEG with a molecular weight of 2000 Da and above gives adequate stealth characteristics.145 Hong et al. reported one of the first attempts to generate a self assembly ABC complex.86 In this instance, DDAB/Chol cationic liposome-based LD particles were stabilised for storage by inclusion of N-[ω-methoxypoly(oxyethylene)-α-oxycarbonyl]-DSPE (PEG-PE) and partially stabilised in the circulation in vivo. However, >1 mol% of PEG-PE proved sufficient to reduce lung in vivo transfection efficacy to a fraction of the transfection level mediated by DDAB/Chol cationic liposomes alone, indicative of a necessary compromise between the requirement to include PEG-PE for stabilisation purposes countered by the requirement to keep levels modest in order prevent “steric blocking” of LD transfection.There are essentially three different ways in which ABC nanoparticles may be formulated with an exterior PEG C-layer (Fig. 9). These are:
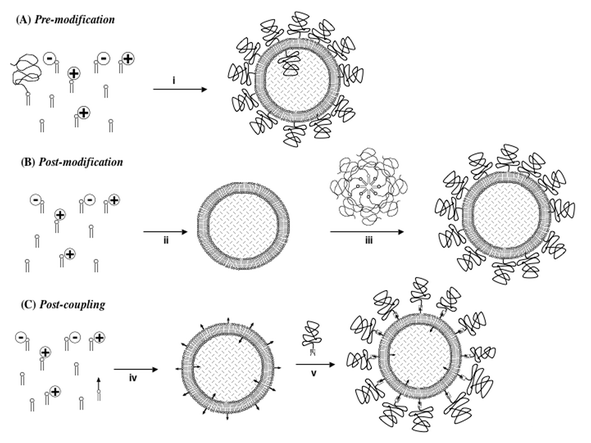 | ||
| Fig. 9 C-layer stealth molecule incorporation strategies. Top:Pre-modification implies that PEG lipids are incorporated into cationic liposomes directly (step i) prior to the addition of any given nucleic acid; Middle:Post-modification strategy implies that simple cationic liposome (or LD, LsiR, LMD or LPD-like) systems are prepared in advance (step ii) and then PEG lipid micelles are incubated (step iii) with the liposome (or LD, LsiR, LMD or LPD-like) particles to encourage micelle breakdown and insertion of PEG lipids via their hydrophobic moieties into the outer leaflet membrane of whichever type of particle is being prepared; Bottom:Post-coupling strategy implies that simple cationic liposome (or LD, LsiR, LMD or LPD-like) systems are formulated with a coupling-lipid that enters lipid membranes (step iv). This coupling-lipid comprises a polar, functional group (black arrow) with very high chemoselectivity for certain complementary functional groups introduced into the termini (checked complementary-arrow shape) of modified PEG-molecules, allowing for highly efficient coupling in the subsequent conjugation step (step v). Post-coupling is flexible enough to allow for the introduction of other biological compatibility/stealth molecules and/or biological targeting molecules (see text for references). (Cationic liposome/micelle–siRNA complex = LsiR.) Reproduced with the kind permission of Elsevier Academic Press.209 | ||
1) Pre-modification: where a PEG-lipid is formulated into cationic liposomes prior to the addition of nucleic acids.86
2) Post-modification: where PEG-lipids in the form of micelles are combined with preformulated AB nanoparticle systems in the expectation that free and micellar PEG-lipids will transfer from free solution or micellar state and insert their hydrophobic lipid moieties into the outer leaflet membranes of AB nanoparticles.98
3) Post-coupling: where PEG-polymers are equipped with reactive functional groups that bioconjugate in aqueous conditions with complementary functional groups presented on the outside surface of the AB nanoparticle.98,102
Free amino functional groups on the surface of LMD particles can be modified easily by post-coupling with a PEG-succinimide activated-ester, giving a simple LMD-based ABC nanoparticle.102 Perhaps surprisingly, these particles were observed to enter cells with ease even though prevailing opinion would have suggested that the PEG stealth layer might provide a steric barrier to cellular uptake. Instead, cellular uptake was found to be rapid (minutes) and substantial, but particles appeared to be entrapped in endosome compartments post cell entry and no measurable transfection was observed.98,102 PEG has undeniable stealth/biocompatibility properties, and clearly facilitates the cellular uptake of attached nanoparticles in spite of these effects. Unfortunately, PEG also appears to block subsequent endosome escape completely. The obvious solution appears to be some form of triggered release of attached PEG once nanoparticles become trapped in endosome compartments revealing naked LMD particles (AB core nanoparticles) that are able to effect endosmolysis and onwards transfection.102 Other polymers such as pluronic acid (a propylene oxide containing block co-polymer) and oligosaccharides also promote DNA uptake into cells.98,146 This appears fundamental. Any given ABC (or even ABCD) system delivering DNA is only likely to be properly clinically viable once triggerable, meaning that they should be completely stable and non-reactive in extracellular fluids but unstable once recognised and/or internalised by target cells in the organ of choice. This paradox goes to the heart of the matter. ABC (or even ABCD) nanoparticles that are not triggerable are unlikely to be effective particles for DNA delivery by their very nature.
In the absence of triggered release, time-dependent release of PEG-lipids has turned out to be a reliable if not entirely effective alternative. According to this approach, PEG-lipids with hydrophobic moieties of variable chain-length will have variable affinities for the outer leaflet membrane of the AB core particle into which they are inserted. The shorter the alkyl chain, the lower will be the affinity and the lower will be the PEG-lipid residence time in the membrane (usually referred to in terms of residence half-life t1/2). Ideally, PEG-lipids should be retained as far as the organ of choice in vivo and even up until the target cells, before dissociation and exposure of naked AB core nanoparticles to enter cells. This feature is characteristic of the rationale leading to the stabilized plasmid-lipid particle (SPLP) systems, the most developed of ABC nanoparticle systems to date. First generation SPLP system contained DOPE 1 (84 mol%), low levels (6 mol%) of cationic lipid dioleyldimethylammonium chloride (DODAC) and quite high levels of a PEG-Ceramide with an arachidoyl acyl group (PEG-CerC20) (10 mol%).147 The surface tenacity of PEG-CerC20 (t1/2 > 13 days) proved such an intractable steric barrier to transfection in vitro that PEG-CerC20 was replaced by PEG-CerC8 (t1/2 < 1.2 min) with an octanoyl acyl group. Entrapment of pDNA was then accomplished by a detergent dialysis procedure (55–70% efficient), giving second-generation DOPE/DODAC/PEG-CerC8 SPLP particles containing DODAC (24–30 mol%) and PEG-CerC8 (15 mol%) (diameter approx. 100 ± 40 nm),148 that were able to effect transfection of cells in vitro and regional delivery of pDNA in vivo. The formulation procedure is illustrated diagrammatically (Fig. 10). One of the most important aspects about SPLP particles is their very structural integrity (no changes in size or DNA encapsulation at 4 °C for 5 months).
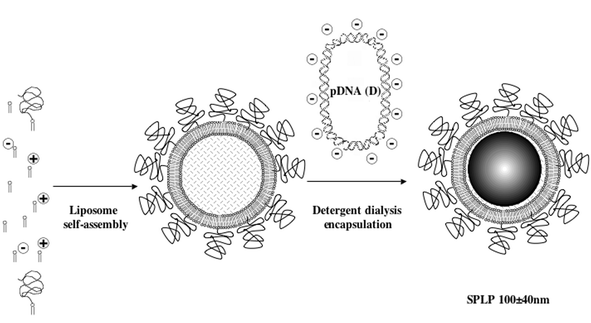 | ||
| Fig. 10 SPLP formulation. Formation of low charge cationic PEG-liposomes by pre-modification with appropriate PEG lipids. The subsequent introduction of condensed pDNA requires some form of detergent analysis (encapsulation efficiency approx. 60%). Excess pDNA must be removed afterwards by column chromatography in order to yield stabilised plasmid-lipid (SPLP) particles ready for use (see text for references). Reproduced with the kind permission of Elsevier Academic Press.209 | ||
SPLP particles were designed for passive targeting. That is to say, particles were designed for long term circulation in vivo enabling the gradual partition of particles into interstitial spaces in diseased tissue (such as tumour) by extravasation from blood via the enlarged sinusoidal gaps that typically exist between the endothelial cells which line the vasculature in diseased and inflamed organs. Given the severity of the extracellular in vivo environment only PEG-CerC20 SPLP particles were found to give modest, detectable transfection in animals post i.v.-administration.149 Other systems such as the PEG-CerC8 SPLP particles were not sufficiently robust to effect passive targeting even though their transfection properties were in principle superior. In other words, in seeking to find the balance between very high extracellular structural integrity (stability) and the very low intracellular or local structural integrity (instability) necessary to promote transfection, the requirement for extracellular stability was too overwhelming with the result that SPLP particles were developed with appropriate extracellular stability but insufficient instability for effective transfection once at the target cells of interest. Improvements continue to be made in order to increase DNA encapsulation efficiency and to improve transfection efficiency.150–153 SPLP systems are now under evaluation in Phase I clinical trials (Protiva, unpublished data), the first ABC nanoparticle to be so evaluated. Data is awaited eagerly. Undoubtedly, SPLP systems represent a key synthetic, non-viral platform technology, and one that others are aiming to emulate increasingly and improve upon.154
The absence of triggered release is still perceived to be the main limitation of SPLP systems. Accordingly, Szoka and coworkers have assembled their own SPLP system taking advantage of a designed ortho ester PEG-lipid known as polyethyleneglycol-diorthoester-distearoyl glycerol conjugate lipid (POD) 21. POD 21 is one of the best bespoke-pH-triggered PEG-lipids to date. The structure and synthesis of POD 21 are shown155,156 (Scheme 7). The key design feature of such a pH-triggered PEG-lipid is that the triggerable linker should be completely stable at pH 7 and sufficiently destabilised at pH 5.5 to completely and irreversibly dissociate within 1 hour at the very least. Such a requirement is severe but is essential to ensure quantitative release of nucleic acid from endosomes! Biophysical release studies were performed with POD-loaded liposomes suggesting that the release half-life, t1/2, at pH 5.5 was approx. 10 min.157 Hence, proof of concept studies were carried out with a POD-SPLP formulation (DOPE/DOTAP/POD 68 ∶ 12 ∶ 20 m/m/m) into which pDNA was encapsulated (40–45% efficiency) by detergent dialysis (as above) giving 60 nm particles. These were found to mediate transfection in vitro much less effectively than simple DOTAP/DOPE cationic liposomes mixed with pDNA. However, POD-SPLP systems were up to three orders of magnitude more effective at transfection than equivalent pH-insensitive nanoparticle systems formulated with PEG-DSG rather than POD 21.158 Both POD-SPLP particles and particles of the equivalent pH-insensitive systems were found to enter cells in line with data obtained with LMD-based ABC nanoparticles.102 Therefore, the clear implication is that the enhanced transfection efficiency of the POD-SPLP system was the result of the triggered release of PEG in the endosome leading to considerably enhanced endosmolysis and pDNA escape to the nucleus. Proof of concept studies in vivo are now awaited, so too are alternative next generation triggerable ABC nanoparticle systems for DNA delivery.
 | ||
| Scheme 7 Reagents and conditions: (i) pTSA, THF, 40 °C; (ii) DCC, DMAP, methoxy-poly(ethyleneglycol)-carboxymethyl (MPEGA).49,155 | ||
Other ABC nanoparticles have been described mostly with regard to triggered or time dependent release properties and provide useful supporting studies. For instance, ABC nanoparticle systems have recently been constructed using short and long-chain SAINT-PEG lipids constructed from pyridinium cytofectins that have variable length residence times in the same way that PEG-ceramide lipids do.159 Alternatively, Thompson and coworkers have adapted their chemical routes to cytofectin BCAT and diplasmenylcholine in order to prepare a novel acid labile PEG-lipid (R)-1,2-di-O-(1′Z, 9′Z-octadecadienyl)-glyceryl-3-(ω-methoxy-poly(ethylene) glycolate 5000) (BVEP) 2249,160,161 (Scheme 7). Elegant though the idea is, biophysical release studies were performed with BVEP -loaded liposomes suggesting that the release half-life, t1/2, at pH 4.5 was approx. 4 hours.160 In this respect, the vinyl ether functional group is probably just too stable to provide for rapid and effective enough triggered release in DNA delivery involving ABC nanoparticles. Otherwise, two other studies are worthy of note. PEG-lipids appear to stabilize LD particles prepared from bis(guanidinium)-tren-cholesterol (BGTC)/DOPE liposomes. Subsequent ABC nanoparticles will transfect murine lung in vivo even without triggered release suggesting that topical lung administration may be exceptional.162 Finally, Scherman and coworkers have been demonstrating how the judicious introduction of anionic PEG moieties can tune the systemic circulation lifetimes of ABC nanoparticles.163,164
6 ABCD particles
For most in vivo applications and gene therapies, true ABCD nanoparticle systems are perhaps the best proposition. The number of these is growing in spite of the obvious technical problems surrounding reproducible and scalable formulation of an AB core particle alongside controlled and reliable association of C and D layer molecules. This remains an ongoing problem. Pardridge and coworkers have reported really impressive results using an ABCD system comprising an LD core particle prepared from cationic liposomes with minimal cytofectin. This LD core is doped with PEG-PE variants, one for stabilisation and one for the covalent attachment of an anti-TfR monoclonal antibody (OX26) specific for TfR that is overexpressed by cells at the blood brain barrier (BBB) and also in peripheral organs such as liver and spleen.165,166 Their main ABCD system is comprised of POPC/DDAB/DSPE-PEG2000 (19.2 ∶ 0.2 ∶ 0.6 m/m/m) liposomes where the DSPE-PEG2000 is distributed DSPE-PEG2000/DSPE-PEG2000-Maleimide (95 ∶ 5 m/m). Particles were prepared by initial mixing of all of the lipids together in chloroform solution followed by solvent evaporation, rehydration, sonication, pDNA addition, concluding with multiple freeze-thaw cycles and extrusion. This arduous process (20% efficient) yielded ABC nanoparticles that were coupled to OX26 antibody overnight, yielding complete ABCD particles (35–50 OX26 MAb/particle; 45–114 nm) after final Sepharose Cl4B gel filtration to remove excess unreacted antibody166 (Fig. 11). | ||
| Fig. 11 PIL formulation. Formation of low charge cationic PEG-liposomes by pre-modification with appropriate PEG lipids. The subsequent introduction of condensed pDNA requires extensive freeze–thaw and extrusion steps (encapsulation efficiency approx. 20%). Excess pDNA must be removed afterwards. Final monoclonal antibody (MAb) coupling then takes place to yield pegylated immunoliposomes (PIL) particles ready for use (see text for references). Reproduced with the kind permission of Elsevier Academic Press.209 | ||
These ABCD nanoparticles are almost completely neutral in charge and do not possess any triggered release system. Therefore, the impressive biological data showing transfection in liver, spleen and brain must be a consequence of active targeting mechanisms involving TfR interactions. From these beginnings, Pardridge and coworkers have developed further the formulation protocols for these pegylated immunoliposome (PIL) systems and demonstrated that PILs have impressively low levels of associated systemic toxicity.167 Additional applications of PILs include the delivery of pDNA-directed epidermal growth factor receptor (EGFR) antisense mRNA using particles “armed” with both an anti-TfR MAb and an anti-insulin receptor (INSR) MAb, the first to promote crossing of the BBB and the second to promote transport of pDNA to the nucleus across plasma and nuclear membranes in the target brain tumour.168,169 Dual targeting was deemed essential to ensure targeting across both the tumour cell membrane and microvasculature barrier to reach cells deep within the cancer tissue. Furthermore, TfR MAb-targeted PILs were used to mediate pDNA-directed short hairpin interference RNA (shRNAi) downregulation of transgenic luciferase activity in vivo in rat cranial brain tumours by up to 90%.170
Others are also moving towards success with the active targeting of tumours after i.v. injection of alternative ABCD systems prepared using MAbs or folate as the targeting ligand.159,171,172 One of the most interesting variations on this theme has been the creation of folate receptor (FR)-targeted DDAB/CHEMS/f-PEG-PE liposomes comprising folate (f) conjugated PEG-PE (f-PEG-PE), cholesteryl hemisuccinate (CHEMS) and the cytofectin dimethyldioctadecylammonium bromide (DDAB) 6, that are combined with poly-L-lysine (pLL) condensed pDNA to give ABCD nanoparticles competent to mediate FR-specific delivery of pDNA to cells in vitro.173 However, whether or not this system will operate effectively in vivo remains to be seen.
7. The future role of biology; A layer innovations
This section is included owing to the fact that most chemists and chemical biologists interested in gene therapy related problems do not pay adequate attention to the nature of the encapsulated nucleic acid (A) that is to be delivered. Normally, chemists and many others assume that once the nucleic acid is “delivered to the nucleus”, then their role is over. However, this is too simplistic. At the very least, the chemist should be aware of the different nucleic acid “payload” possibilities that are available for delivery. At the very best, the chemist should not only have a feel for the differences in molecular biology that govern their mechanisms of action of each alternative payload, but appreciate how synthetic non-viral vector systems could be adapted or fitted out for different nucleic acid payloads according to those mechanisms of action. Indeed, some synthetic non-viral vectors will also be better adapted naturally for the delivery of some nucleic acid payloads compared with others. For instance, while LD systems that tend to form metastable, large particles (>150 nm) appear to be well adapted for pDNA delivery in vitro,38 LD systems with a membrane active component that generate small, stable particles (<150 nm) appear to be better adapted for the efficient delivery of anti-sense phosphorothiolate oligonucleotides in vitro.1747.1 DNA constructs
Broadly speaking, payloads will either be DNA or RNA in character. In the case of DNA, pDNA has been the most commonly used form of DNA and has also been the most commonly used form of nucleic acid. Much of the data described in the preceding chapters has been acquired using pDNA. DNA must always be delivered to the nucleus in order to demonstrate a function, as described previously, but problems are far from over once there. Typically, gene expression takes place post-nuclear delivery and declines to background levels between 7 and 14 days post-transfection. This is known as plasmid silencing. Plasmid silencing is unhelpful for most projected in vivo applications or gene therapy. Curiously, the reasons for plasmid silencing do not appear to be plasmid shedding (loss of pDNA from cells) or plasmid CpG methylation as might be expected,175 suggesting that other mechanisms are involved such as chromatin remodelling (nuclear protein condensation of pDNA leading to inactivation of gene expression). While research into plasmid silencing would be of undoubted use in the design of long-term expression plasmids such as plasmid minicircles,176 molecular biology has not stood still and a number of alternatives to simple epichromosomal pDNA now exist that could be used in place.A potential way to enhance long term expression in pDNA may be to introduce elements of DNA structure involved in the control of gene expression. Obviously, just as open-reading frames (ORFs) (genes or sections of genes) only comprise a fraction of chromosomal DNA in any one cell so open-reading frames do not comprise the entirety of any one plasmid. There are now known to be elements such as Locus Control Regions (LCRs) and Ubiquitous Chromatin Opening Elements (UCOEs) that act to sustain associated ORFs in states appropriate for transcription (into mRNA) and promote long term expression.177 These have proven themselves in viral vector systems and now await analysis in synthetic non-viral vector systems.
Otherwise, the arrival of mammalian transposons (transposable elements) now looks remarkably promising. Transposons are stretches of DNA (either linear or circular) that are capable of insertion into chromosomal DNA at defined sites with the assistance of a transposase enzyme.178 Of particular significance is the Sleeping Beauty transposon element originally identified in the Zebra fish genome through sequence similarity with active transposons (Tc1/mariner transposable elements) found in Drosophila and Caenorhabditis elegans but rendered inactive in Zebra fish through deleterious mutations. Reversal of these mutations has created a transposon active in mammalian cells and able to insert a transgene embedded in the transposon sequence into mammalian chromosomal DNA leading to long term transgene expression (months)179–181 (Fig. 12). Proof of principle studies with non-viral delivery of Sleeping Beauty transposon to cells have been accomplished,182 although there are concerns that this transposon integrates into too many sites in chromosomal DNA and therefore may be cancer-inducing (oncogenic) in the same way that retroviridae are. The physico-chemical properties of Sleeping Beauty transposon integration sites in chromosomal DNA are known (palindromic AT repeat) and are theoretically numerous.183 However, transposase enzymes do not integrate transposons into genes under active transcription unlike retroviridae. Therefore, the risks of oncogenicity are much reduced but remain realistic at this stage. Accordingly, there have been proposals for the construction of chimeric-transposase enzymes engineered with binding domains (such as zinc-finger domain proteins) with high affinity for select DNA sequences that could guide the transposon to site specific integration.184 This idea is seductive but requires complete validation. An alternative approach has been suggested and validated using an integrase enzyme ϕC31 from bacteriophage. In this instance, the enzyme integrates an alternative transposable element that interacts with chromosomal DNA at binding sites less prevalent than the Sleeping Beauty integration sites and consequently is perceived to minimise the risks of oncogenicity in comparison185–188 (Fig. 13). This approach is still in the early stages of technical development but appears potentially useful.
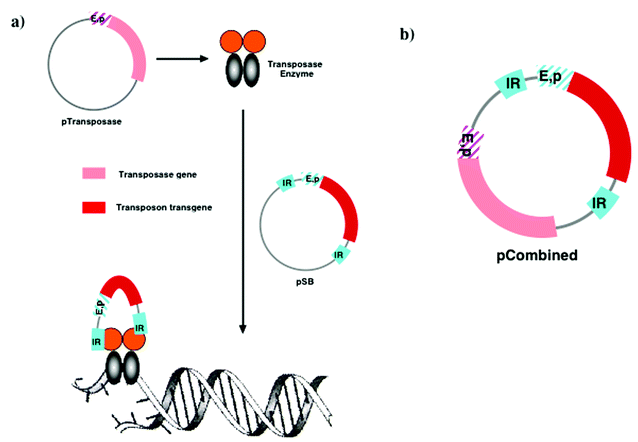 | ||
| Fig. 12 Outline mechanism of Sleeping Beauty transposon mechanism. (a) Schematic to show how dimeric sleeping beauty transposase is coded for by one pDNA (pTransposase) and how this expressed enzyme then captures the inverted repeats (IRs) found in the pDNA (pSB) harbouring the transposon. Dimeric transposase then excises the complete transposon and relocates to defined chromosomal DNA binding sites where insertion of the complete transposon is finally assisted (see text for references); (b) Schematic to show how transposon and transposase gene could be integrated together on the same pDNA. | ||
 | ||
| Fig. 13 Outline mechanism of phage integrase mechanism. Schematic to show how attP DNA sites in a given pDNA recognise and interact with complementary attB sites in chromosomal DNA. Thereafter, phage integrase enzyme ϕC31 performs insertion of the entire pDNA into the attB sites in a unidirectional and robust manner without the need for cofactors (see text for references). The same will be true when attB sites are located in pDNA and interact with attP-like sites in chromosomal DNA. | ||
Artificial chromosomes represent the main alternative to pDNA and may be delivered with synthetic non-viral vectors to cells owing to the fact that these vector systems are able to deliver any nucleic acid construct irrespective of size. Clearly artificial chromosomes (50 kbp–1 Mbp) are much larger than pDNA (typically 4–7 kbp) but have been constructed to express genes in an epichomosomal manner supported by all the main features of a chromosome (such as the centromere) so that they operate as pseudo-chromosomes. Depending upon source of sequences and genes, there are bacterial artificial chromosomes (BACs),189–191 P1-derived artificial chromosomes (PACs),189,192,193 yeast artificial chromosomes (YACs),194,195 mammalian artificial chromosomes (MACs),196–198 and human artificial chromosomes (huACs).199,200 Proof of concept has been demonstrated using synthetic non-viral vector systems and artificial chromosomes resulting in long term expression in cells in vitro,189,192,198 and even in vivo.190,191 At the other end of the spectrum, DNA aptamers and ribozyme DNAs have also been delivered successfully to cells in vitro. HIV-1 gene expression was successfully inhibited by the intervention of anti-HIV Rev-binding aptamer [RBE(apt)], and a ribozyme directed against the HIV-1 env gene,201 both delivered by an ABD nanoparticle.
7.2 RNA constructs
In the case of RNA, one might consider the delivery of mRNA but the complex, heterogeneous secondary structure of such molecules and perceived vulnerability to hydrolysis have ensured that mRNA has seen little application. However, the delivery of siRNA is set to transform the use of RNA and potentially even the face of therapeutic medicine itself. The concept of siRNA has risen with incredible speed within the past two years.202 The phenomenon of RNA interference (RNAi) has a provenance stretching back to at least 1995 when large double stranded RNAs (dsRNA) were found to silence genes in nematodes by a mechanism that is only now being properly appreciated (Fig. 14). According to this mechanism, dsRNA is broken down into siRNA duplexes (typically comprising 2 base overhangs at each 3′-end and a central antiparallel 19bp double helical region) by an enzyme system known as DICER. The siRNAs then associate with a protein complex (RISC) that interacts in an asymmetric manner with each siRNA, separating sense (S) and antisense (AS) strands from each other and preferentially adopting the AS over the S strand as a “template” to bind target mRNA. Target mRNA is singled out for destruction by this activated RISC through siRNA-mediated target recognition apparently provided by the AS strand of siRNA bound to RISC that presumably makes complementary Watson–Crick base pair interactions with a corresponding region in target mRNA.203,204 | ||
| Fig. 14 Outline of siRNA mechanism of action. Small interference RNA (siRNA) is derived from long double-stranded RNA (dsRNA) through the action of the DICER enzyme system. Interaction of siRNA molecules with the RISC enzyme system results in sense (S)/anti-sense (AS) strand separation and the probable capture of individual AS strands by RISC. So activated, RISC recognises mRNA molecules with Watson–Crick base-pair complementary to bound AS strand. Once recognised and bound, mRNA is cleaved and then degraded.202 | ||
What makes siRNA so potent is that numbers of appropriate siRNAs may be identified with the capacity to complement corresponding regions in a target mRNA of interest, after which these siRNAs may be sifted with defined rule sets so as to identify those siRNAs (approx. 1–3% per gene) with S and AS strand base sequences that are optimal for RISC-mediated destruction of the target mRNA of interest.205 Furthermore, these sequences may be screened at one higher level by means of high-end bioinformatics analyses (such as the siDIRECT analysis) ensuring that they have no likelihood of cross-reactivity with other off-target mRNA sequences and hence little likelihood of eliciting undesirable cellular toxicities.206 The role of siRNA in genomics and target validation through specific gene knockdown and phenotypic characterisation is now beyond question. Moreover, with such apparent precision, a role of siRNA therapeutics now seems very credible. Gratifyingly, cationic liposome mediated siRNA delivery to cells (siFection) is particularly suitable for siRNA applications, not the least because cationic liposome systems such as CDAN/DOPE (45 ∶ 55 m/m, siFECTamine®) have been specially formulated and adapted for in vitro siFection of cells resulting in maximum gene knockdown efficacy (>90%) with absolutely minimal toxicity.39 The siFECTamine® cationic liposome system can be upgraded in a modular fashion for in vivo siFection (see below). Clearly, successful siFection does not require siRNA delivery to cell nuclei and also there is no real equivalent to the “long term expression” problem with pDNA. Therefore effective delivery of siRNA in vivo looks to be much more straightforward to achieve than effective delivery of pDNA. Hence the future for in vivo applications of siRNA looks bright not to mention the possibility of siRNA therapeutics as well.
8. The future role of chemistry; B, C and D layer innovations
Previously, we have noted that platform technologies like LMD and SPLP systems should be the only meaningful way forward for cationic liposome/micelle-based systems for in vivo applications and gene therapy. Our reasons for stating this were that these systems represent well-characterised transfection vehicles constructed from tool-kits of well-defined chemical components, that can be formulated in a reproducible and scalable manner, giving rise to reproducible transfection outcomes. We would now like to take the opportunity to update our comments in the light of the self-assembly ABCD nanoparticle concept presented in this review. Hence, in our revised view the most appropriate way forward for cationic liposome/micelle-based synthetic non-viral vector systems, is now the creation of ABC and ABCD nanoparticle systems that have been self-assembled in a modular and sequential fashion from tool-kits of well-defined chemical components. These systems must formulate with nucleic acids of choice in a reproducible and scalable manner giving discrete and well-defined particles with a narrow particle size-distribution in solution (i.e., narrow polydispersity) centred around 70–100 nm diameter, thereby being too large for rapid excretion and too small for rapid RES clearance. Furthermore, these particles should be able to give reproducible transfection outcomes (or siFection outcomes, as appropriate) in vivo with minimal toxicity. The combination of all these features should ensure regulatory confidence in the therapeutic applications of such ABC and ABCD nanoparticle systems. Furthermore, the very self-assembly and modular build characteristics of such nanoparticle systems should ensure that nanoparticles can be tailor-made for individual nucleic acid delivery requirements by using a range of different tool-kits of well-defined chemical components. It is in the synthesis and integration of these tool-kits of chemical components that the future opportunity for chemistry now lies.As stated above, the cytofectin (B-layer components) field appears to be approaching saturation. There are numerous examples of cytofectins in the literature and the field of non-viral gene therapy is now less unlikely to benefit from further additions. However, there is real benefit in the synthesis of new lipids to facilitate the attachment and function of the stealth/biocompatibility C-layer. For instance, lipids containing the aminoxy functional group shown above (Scheme 5) can be synthesised, formulated into complexes with nucleic acids and then used to effect the post-coupling of PEG-aldehydes in aqueous medium. The result of this aqueous post-coupling procedure appears to be the highly efficient, reliable and non-disruptive introduction of a biocompatible/stealth C-layer resulting in robust ABC/ABCD nanoparticles, such as siFECTplus™ nanoparticles for the functional delivery of siRNA to cells in organs in vivo (see CONZENTRx™ systems of IC-Vec Ltd, unpublished results). Post coupling through aminoxy functional groups is clearly potentially effective, but what of other functional groups? Alternatively, requirements for B and C layer innovation could be combined in the quest for alternatives to the pH-triggerable PEG-lipids of Szoka or Thompson and coworkers (Scheme 7). The recent review of Guo and Szoka207 was compiled not only to illustrate pH triggering but also redox potential, temperature and even enzymatic triggering processes. There is plenty of room for chemical innovation here!
Then there is the question of PEG itself. This remains the mainstay for most in vivo applications involving viable ABC and ABCD nanoparticles. However, this large and unwieldy “stealth/biocompatibility” molecule has already been shown to be refractory for transfection with pDNA.98,102 Triggered release of PEG from the AB core once nanoparticles have entered cells, seems imperative in order for effective pDNA transfection to take place,158 although the presence of attached PEG may in fact be much less a problem for siFection (delivery of siRNA). Nevertheless, efforts should be put into finding alternative hydrophilic polymers that can mimic the biocompatibility and stealth properties of PEG without the refractory characteristics and lack of biodegradability. Some alternative hydrophilic polymers have already been described by Seymour and coworkers, including poly-[N-(2-hydroxypropyl)methacrylamide] (pHMPA).208 Once again, there should be a host of other possibilities once the enthusiasm of polymer chemists can be engaged on this problem. Once prepared, each prospective surrogate of PEG will have to be rigorously evaluated for biocompatibility and stealth properties coupled with low toxicity and adequate biodegradability. However, this process should be encouraged at the earliest opportunity.
In general, there is a real requirement for more and better bioconjugation methodologies for the coupling of biological targeting moieties to core AB or ABC nanoparticles. Thus far, bioconjugation methodologies have been few and rather inefficient, including the aqueous coupling between free thiol groups and maleimide functional groups, or free amino groups and succinimide-activated esters. Moreover, there is usually little effort to characterise and confirm the results of most bioconjugation reactions that have been described in the literature, and correspondingly little real effort to separate bioconjugation products from reactants! This is woeful and also needs addressing at the earliest opportunity. Aminoxy-aldehyde aqueous functional group coupling and robust high performance liquid chromatography (HPLC) analyses have recently been developed (IC-Vec Ltd, unpublished results) and appear to represent an effective means of D-layer bioconjugation. However, once again much more chemical diversity is required for reproducible and scalable aqueous coupling of peptides, proteins and/or oligosaccharide targeting moieties to core AB or ABC nanoparticles.
By way of final comment, the circulatory extracellular barriers discussed in Section 1.2.1 of this review were said to be far from exhaustive and are primarily valid as long as synthetic non-viral vector systems are involved in local delivery applications in vivo to lung, peritoneal cavity, vascular system or main filtration organs such as the liver. For systemic delivery to other organs including tumours, there are potentially other significant issues concerning tissue penetration, cell organisation, and access to cells of interest through the extracellular matrix that have not been addressed substantially in this review. These additional barriers are only just beginning to be thought about for synthetic non-viral vector systems and may be insurmountable. However, this will not become clear until extensive pharmacokinetic studies can be carried out with radioactive or specifically fluorescent-labelled ABC or ABCD nanoparticles. Therefore, chemical synthesis of bespoke probes for multiply-labelled nanoparticles,102 is yet another area of chemical synthesis and innovation that could contribute significantly to synthetic non-viral vector gene therapy going forward, used in combination with in vivo studies and increasingly sophisticated in vitro/ex vivo cell model systems (spheroids and 3D cellular multilayers) designed to study these additional barriers to successful transfection in isolation.
Main abbreviations
DOPE, dioleoyl-L-α-phosphatidylethanolamineChol, cholesterol
DOTMA, N-[1-(2,3-dioleyloxy)propyl]-N,N,N-trimethyl ammonium chloride
DOTAP, 1,2-dioleoyloxy-3-(trimethylammonio)propane
DOSPA, 2,3-dioleyloxy-N-[2-(sperminecarboxamido)ethyl]-N,N-dimethyl-1-propanaminium trifluoroacetate
DDAB, dimethyldioctadecylammonium bromide
DOGS, dioctadecylamidoglycylspermine
DC-Chol, 3β-[N-(N′,N′-dimethylaminoethane)carbamoyl]cholesterol
CDAN, N1-cholesteryloxycarbonyl-3,7-diazanonane-1,9-diamine
BGTC, bis-guanidinium-tren-cholesterol
DOTIM, 1-[2-(oleoyloxy)ethyl]-2-oleyl-3-(2-hydroxyethyl)imidazolinium chloride
SAINT, Synthetic Amphiphiles INTerdisciplinary
TMAG, N-(α-trimethylammonioacetyl)-didodecyl-D-glutamate chloride
BCAT, O-(2R-1,2-di-O-(1′Z,9′Z-octadecadienyl)-glycerol)-N-(bis-2-aminoethyl)carbamate
GS11, Gemini Surfactant 11
5AMyr, 1-(1,3-dimyristoyloxypropane-2-yl)-2,4,6-trimethylpyridinium hexafluorophosphate
3AMyr, 1-(2,3-dimyristoyloxypropyl)-2,4,6-trimethylpyridinium hexafluorophosphate
DODAC, dioleyldimethylammonium chloride
PEG-PE, N-[ω-methoxypoly(oxyethylene)-α-oxycarbonyl]-DSPE
PEG, polyethylene glycol
DSPE, distearoyl-L-α-phosphatidylethanolamine
PEG-CerC20, PEG-Ceramide bioconjugate with an arachidoyl acyl group
PEG-CerC8, PEG-Ceramide bioconjugate with an octanoyl acyl group
POD, polyethyleneglycol-diorthoester-distearoyl glycerol conjugate
DSG, distearoyl glycerol
BVEP, (R)-1,2-di-O-(1′Z,9′Z-octadecadienyl)-glyceryl-3-(ω-methoxy-poly(ethylene) glycolate 5000)
pHMPA, poly-[N-(2-hydroxypropyl)methacrylamide]
CHEMS, cholesteryl hemisuccinate
pLL, poly-L-lysine
LD, lipoplex (cationic liposome/micelle–DNA complex)
LsiR, cationic liposome/micelle–siRNA complex
LPD, liposome:polycation:DNA (lipid:protamine:DNA)
LMD, liposome:mu:DNA
mu, μ peptide (of adenovirus)
MEND, multifunctional envelope-type nano device
STR-R8, stearyl octaarginine
SPLP, stabilized plasmid-lipid particles
HVJ, hemagglutinating virus of Japan (Sendai virus)
PIL, pegylated immunoliposome
ODN, oligodeoxynucleotide
ON, oligonucleotide
pDNA, plasmid DNA
siRNA, small interference RNA
shRNAi, short hairpin interference RNA
BAC, bacterial artificial chromosome
YAC, yeast artificial chromosome
MAC, mammalian artificial chromosome
PAC, P1-derived artificial chromosome
huAC, human artificial chromosome
RES, reticulo-endothelial system
Tf, transferrin
TfR, transferrin receptor
EGFR, epidermal growth factor receptor
INSR, insulin receptor
f, folate
FR, folate receptor
References
- A. D. Miller, Global Outsourcing Review, 2002, 4, 8 Search PubMed.
- C. Kitson, B. Angel, D. Judd, S. Rothery, N. J. Severs, A. Dewar, L. Huang, S. C. Wadsworth, S. H. Cheng, D. M. Geddes and E. W. Alton, Gene Ther., 1999, 6, 534 CrossRef CAS.
- D. C. Litzinger, J. M. Brown, I. Wala, S. A. Kaufman, G. Y. Van, C. L. Farrell and D. Collins, Biochim. Biophys. Acta, 1996, 1281, 139.
- F. Liu, H. Qi, L. Huang and D. Liu, Gene Ther., 1997, 4, 517 CrossRef CAS.
- S. Li, W. C. Tseng, D. B. Stolz, S. P. Wu, S. C. Watkins and L. Huang, Gene Ther., 1999, 6, 585 CrossRef CAS.
- J. P. Yang and L. Huang, Gene Ther., 1997, 4, 950 CrossRef CAS.
- J. P. Yang and L. Huang, Gene Ther., 1998, 5, 380 CrossRef CAS.
- O. Zelphati, L. S. Uyechi, L. G. Barron and F. C. Szoka, Jr., Biochim. Biophys. Acta, 1998, 1390, 119 CAS.
- C. Plank, K. Mechtler, F. C. Szoka, Jr. and E. Wagner, Hum. Gene Ther., 1996, 7, 1437 CAS.
- S. W. Dow, L. G. Fradkin, D. H. Liggitt, A. P. Willson, T. D. Heath and T. A. Potter, J. Immunol., 1999, 163, 1552 CAS.
- S. Li, S. P. Wu, M. Whitmore, E. J. Loeffert, L. Wang, S. C. Watkins, B. R. Pitt and L. Huang, Am. J. Physiol., 1999, 276, L796 Search PubMed.
- G. McLachlan, B. J. Stevenson, D. J. Davidson and D. J. Porteous, Gene Ther., 2000, 7, 384 CrossRef CAS.
- S. Pasquini, H. Deng, S. T. Reddy, W. Giles-Davis and H. C. Ertl, Gene Ther., 1999, 6, 1448 CrossRef CAS.
- F. G. Zhu, C. F. Reich and D. S. Pisetsky, Immunology, 2003, 109, 255 CrossRef CAS.
- G. Osaka, K. Carey, A. Cuthbertson, P. Godowski, T. Patapoff, A. Ryan, T. Gadek and J. Mordenti, J. Pharm. Sci., 1996, 85, 612 CrossRef CAS.
- Y. K. Song, F. Liu, S. Chu and D. Liu, Hum. Gene Ther., 1997, 8, 1585 CAS.
- H. E. Hofland, D. Nagy, J. J. Liu, K. Spratt, Y. L. Lee, O. Danos and S. M. Sullivan, Pharm. Res., 1997, 14, 742 CrossRef CAS.
- L. C. Mounkes, W. Zhong, G. Cipres-Palacin, T. D. Heath and R. J. Debs, J. Biol. Chem., 1998, 273, 26164 CrossRef CAS.
- A. D. Miller, Angew. Chem. Int. Ed., 1998, 37, 1768 CrossRef.
- A. D. Miller, Curr. Res. Mol. Ther., 1998, 1, 494 Search PubMed.
- A. D. Miller, Understanding Gene Therapy, ed. N. R. Lemoine, Bios Scientific Publishers, 1999 Search PubMed.
- W. C. Tseng, F. R. Haselton and T. D. Giorgio, Biochim. Biophys. Acta, 1999, 1445, 53 CrossRef CAS.
- S. R. Wente, Science, 2000, 288, 1374 CrossRef CAS.
- A. I. Aronsohn and J. A. Hughes, J. Drug Target., 1998, 5, 163 Search PubMed.
- C. Neves, V. Escriou, G. Byk, D. Scherman and P. Wils, Cell Biol. Toxicol., 1999, 15, 193 CrossRef CAS.
- A. Subramanian, P. Ranganathan and S. L. Diamond, Nat. Biotechnol., 1999, 17, 873 CrossRef CAS.
- M. A. Zanta, P. Belguise-Valladier and J. P. Behr, Proc. Natl. Acad. Sci. USA, 1999, 96, 91 CrossRef CAS.
- D. Lechardeur, K. J. Sohn, M. Haardt, P. B. Joshi, M. Monck, R. W. Graham, B. Beatty, J. Squire, H. O'Brodovich and G. L. Lukacs, Gene Ther., 1999, 6, 482 CrossRef CAS.
- A. Wilber, G. Armour and M. C. Schneider, Mol. Ther., 2004, 9, 474.
- G. L. Lukacs, P. Haggie, O. Seksek, D. Lechardeur, N. Freedman and A. S. Verkman, J. Biol. Chem., 2000, 275, 1625 CrossRef CAS.
- E. Dauty and A. S. Verkman, Mol. Ther., 2004, 9, 678.
- M. A. Ilies and A. T. Balaban, Expert Opin. Ther. Patents, 2001, 11, 1729 Search PubMed.
- A. D. Miller, Curr. Med. Chem., 2003, 10, 1195 CrossRef CAS.
- C. Nicolazzi, M. Garinot, N. Mignet, D. Scherman and M. Bessodes, Curr. Med. Chem., 2003, 10, 1263 CrossRef CAS.
- R. G. Cooper, C. J. Etheridge, L. Stewart, J. Marshall, S. Rudginsky, S. H. Cheng and A. D. Miller, Chem. Eur. J., 1998, 4, 137 CrossRef.
- L. Stewart, M. Manvell, E. Hillery, C. J. Etheridge, R. G. Cooper, H. Stark, M. van-Heel, M. Preuss, E. W. F. W. Alton and A. D. Miller, J. Chem. Soc., Perkin Trans. 2, 2001, 624 RSC.
- T. Tagawa, M. Manvell, N. Brown, M. Keller, E. Perouzel, K. D. Murray, R. P. Harbottle, M. Tecle, F. Booy, M. C. Brahimi-Horn, C. Coutelle, N. R. Lemoine, E. W. Alton and A. D. Miller, Gene Ther., 2002, 9, 564 CrossRef CAS.
- M. Keller, M. R. Jorgensen, E. Perouzel and A. D. Miller, Biochemistry, 2003, 42, 6067 CrossRef CAS.
- S. Spagnou, A. D. Miller and M. Keller, Biochemistry, 2004, 43, 13348 CAS.
- M. Oliver, M. R. Jorgensen and A. D. Miller, Tetrahedron Lett., 2004, 45, 3105 CrossRef CAS.
- M. A. Ilies, W. A. Seitz, M. T. Caproiu, M. Wentz, R. E. Garfield and A. T. Balaban, Eur. J. Org. Chem., 2003, 2645 CrossRef CAS.
- M. A. Ilies, W. A. Seitz, I. Ghiviriga, B. H. Johnson, A. Miller, E. B. Thompson and A. T. Balaban, J. Med. Chem., 2004, 47, 3744 CrossRef CAS.
- I. van der Woude, A. Wagenaar, A. A. Meekel, M. B. ter Beest, M. H. Ruiters, J. B. Engberts and D. Hoekstra, Proc. Natl. Acad. Sci. USA, 1997, 94, 1160 CrossRef CAS.
- I. Solodin, C. S. Brown, M. S. Bruno, C. Y. Chow, E. H. Jang, R. J. Debs and T. D. Heath, Biochemistry, 1995, 34, 13537 CrossRef CAS.
- C. McGregor, C. Perrin, M. Monck, P. Camilleri and A. J. Kirby, J. Am. Chem. Soc., 2001, 123, 6215 CrossRef CAS.
- A. J. Kirby, P. Camilleri, J. B. F. N. Engberts, M. C. Feiters, R. J. M. Nolte, O. Soderman, M. Bergsma, P. C. Bell, M. L. Fielden, C. L. G. Rodriguez, P. Guedat, A. Kremer, C. McGregor, C. Perrin, G. Ronsin and M. C. P. van Eijk, Angew. Chem. Int. Ed., 2003, 42, 1448 CrossRef CAS.
- J. A. Boomer, D. H. Thompson and S. M. Sullivan, Pharm. Res., 2002, 19, 1292 CrossRef CAS.
- J. Shin and D. H. Thompson, J. Org. Chem., 2003, 68, 6760 CrossRef CAS.
- J. A. Boomer and D. H. Thompson, Chem. Phys. Lipids, 1999, 99, 145 CrossRef CAS.
- J. S. Remy, A. Kichler, V. Mordvinov, F. Schuber and J. P. Behr, Proc. Natl. Acad. Sci. USA, 1995, 92, 1744 CAS.
- J. P. Behr, B. Demeneix, J. P. Loeffler and J. Perez-Mutul, Proc. Natl. Acad. Sci. USA, 1989, 86, 6982 CAS.
- J. H. Felgner, R. Kumar, C. N. Sridhar, C. J. Wheeler, Y. J. Tsai, R. Border, P. Ramsey, M. Martin and P. L. Felgner, J. Biol. Chem., 1994, 269, 2550 CAS.
- E. W. F. W. Alton, P. G. Middleton, N. J. Caplen, S. N. Smith, D. M. Steel, F. M. Munkonge, P. K. Jeffery, D. M. Geddes, S. L. Hart, R. Williamson, K. I. Fasold, A. D. Miller, P. Dickinson, B. J. Stevenson, G. McLachlan, J. R. Dorin and D. J. Porteous, Nat. Genet., 1993, 5, 135 CrossRef.
- A. McQuillin, K. D. Murray, C. J. Etheridge, L. Stewart, R. G. Cooper, P. M. Brett, A. D. Miller and H. M. Gurling, Neuroreport, 1997, 8, 1481 Search PubMed.
- K. Fife, M. Bower, R. G. Cooper, L. Stewart, C. J. Etheridge, R. C. Coombes, L. Buluwela and A. D. Miller, Gene Ther., 1998, 5, 614 CrossRef CAS.
- K. K. Son, D. H. Patel, D. Tkach and A. Park, Biochim. Biophys. Acta, 2000, 1466, 11 CrossRef CAS.
- F. Labat-Moleur, A. M. Steffan, C. Brisson, H. Perron, O. Feugeas, P. Furstenberger, F. Oberling, E. Brambilla and J. P. Behr, Gene Ther., 1996, 3, 1010 CAS.
- J. Zabner, A. J. Fasbender, T. Moninger, K. A. Poellinger and M. J. Welsh, J. Biol. Chem., 1995, 270, 18997 CrossRef CAS.
- J. O. Radler, I. Koltover, T. Salditt and C. R. Safinya, Science, 1997, 275, 810 CrossRef CAS.
- D. D. Lasic, H. Strey, M. C. A. Stuart, R. Podgornik and P. M. Frederik, J. Am. Chem. Soc., 1997, 119, 832 CrossRef CAS.
- J. Gustafsson, G. Arvidson, G. Karlsson and M. Almgren, Biochim. Biophys. Acta, 1995, 1235, 305 CrossRef CAS.
- H. Gershon, R. Ghirlando, S. B. Guttman and A. Minsky, Biochemistry, 1993, 32, 7143 CrossRef CAS.
- S. W. Hui, M. Langner, Y. L. Zhao, P. Ross, E. Hurley and K. Chan, Biophys. J., 1996, 71, 590 CAS.
- S. J. Eastman, C. Siegel, J. Tousignant, A. E. Smith, S. H. Cheng and R. K. Scheule, Biochim. Biophys. Acta, 1997, 1325, 41 CrossRef CAS.
- B. Sternberg, F. L. Sorgi and L. Huang, FEBS Lett., 1994, 356, 361 CrossRef CAS.
- M. Schmutz, D. Durand, A. Debin, Y. Palvadeau, A. Etienne and A. R. Thierry, Proc. Natl. Acad. Sci. USA, 1999, 96, 12293 CrossRef CAS.
- Y. Xu, S. W. Hui, P. Frederik and F. C. Szoka, Jr., Biophys. J., 1999, 77, 341 CAS.
- B. Pitard, N. Oudrhiri, J. P. Vigneron, M. Hauchecorne, O. Aguerre, R. Toury, M. Airiau, R. Ramasawmy, D. Scherman, J. Crouzet, J. M. Lehn and P. Lehn, Proc. Natl. Acad. Sci. USA, 1999, 96, 2621 CrossRef CAS.
- C. L. Densmore, T. H. Giddings, J. C. Waldrep, B. M. Kinsey and V. Knight, J. Gene Med., 1999, 1, 251 CrossRef CAS.
- I. Koltover, T. Salditt, J. O. Radler and C. R. Safinya, Science, 1998, 281, 78 CrossRef CAS.
- B. Mui, Q. F. Ahkong, L. Chow and M. J. Hope, Biochim. Biophys. Acta, 2000, 1467, 281 CAS.
- J.-P. Behr, M/S-Med. Sci., 1996, 12, 56 Search PubMed.
- J. Haensler and F. C. Szoka, Jr., Bioconjugate Chem., 1993, 4, 372 CrossRef CAS.
- M. E. Ferrari, D. Rusalov, J. Enas and C. J. Wheeler, Nucleic Acids Res., 2002, 30, 1808 CrossRef CAS.
- F. L. Sorgi, S. Bhattacharya and L. Huang, Gene Ther., 1997, 4, 961 CrossRef CAS.
- S. Li and L. Huang, Gene Ther., 1997, 4, 891 CrossRef CAS.
- S. Li, M. A. Rizzo, S. Bhattacharya and L. Huang, Gene Ther., 1998, 5, 930 CrossRef CAS.
- B. Li, S. Li, Y. Tan, D. B. Stolz, S. C. Watkins, L. H. Block and L. Huang, J. Pharm. Sci., 2000, 89, 355 CrossRef CAS.
- M. Whitmore, S. Li and L. Huang, Gene Ther., 1999, 6, 1867 CrossRef CAS.
- S. Chesnoy and L. Huang, Annu. Rev. Biophys. Biomol. Struct., 2000, 29, 27 CrossRef CAS.
- S. Dokka, D. Toledo, X. Shi, J. Ye and Y. Rojanasakul, Int. J. Pharm., 2000, 206, 97 CrossRef CAS.
- J. C. Birchall, I. W. Kellaway and M. Gumbleton, Int. J. Pharm., 2000, 197, 221 CrossRef CAS.
- S. Mizuarai, K. Ono, J. You, M. Kamihira and S. Iijima, J. Biochemistry, 2001, 129, 125 Search PubMed.
- X. Gao and L. Huang, Biochemistry, 1996, 35, 1027 CrossRef CAS.
- L. Vitiello, A. Chonn, J. D. Wasserman, C. Duff and R. G. Worton, Gene Ther., 1996, 3, 396 CAS.
- K. Hong, W. Zheng, A. Baker and D. Papahadjopoulos, FEBS Lett., 1997, 400, 233 CrossRef CAS.
- X. Zhou and L. Huang, Biochim. Biophys. Acta, 1994, 1189, 195 CAS.
- J. D. Fritz, H. Herweijer, G. Zhang and J. A. Wolff, Hum. Gene Ther., 1996, 7, 1395 CAS.
- Y. Namiki, T. Takahashi and T. Ohno, Gene Ther., 1998, 5, 240 CrossRef CAS.
- B. Schwartz, M. A. Ivanov, B. Pitard, V. Escriou, R. Rangara, G. Byk, P. Wils, J. Crouzet and D. Scherman, Gene Ther., 1999, 6, 282 CrossRef CAS.
- L. Vaysse and B. Arveiler, Biochim. Biophys. Acta, 2000, 1474, 244 CrossRef CAS.
- Q. R. Chen, L. Zhang, S. A. Stass and A. J. Mixson, Gene Ther., 2000, 7, 1698 CrossRef CAS.
- C. W. Anderson, M. E. Young and S. J. Flint, Virology, 1989, 172, 506 CAS.
- K. Hosokawa and M. T. Sung, J. Virol., 1976, 17, 924 CAS.
- M. Preuss, M. Tecle, I. Shah, D. A. Matthews and A. D. Miller, Org. Biomol. Chem., 2003, 1, 2430 RSC.
- M. Tecle, M. Preuss and A. D. Miller, Biochemistry, 2003, 42, 10343 CrossRef CAS.
- E. R. Lee, J. Marshall, C. S. Siegel, C. Jiang, N. S. Yew, M. R. Nichols, J. B. Nietupski, R. J. Ziegler, M. B. Lane, K. X. Wang, N. C. Wan, R. K. Scheule, D. J. Harris, A. E. Smith and S. H. Cheng, Hum. Gene Ther., 1996, 7, 1701 CAS.
- E. Perouzel, M. R. Jorgensen, M. Keller and A. D. Miller, Bioconjugate Chem., 2003, 14, 884 CrossRef CAS.
- A. Kichler, K. Mechtler, J. P. Behr and E. Wagner, Bioconjugate Chem., 1997, 8, 213 CrossRef CAS.
- H. Kamata, H. Yagisawa, S. Takahashi and H. Hirata, Nucleic Acids Res., 1994, 22, 536 CAS.
- D. C. Drummond, M. Zignani and J. Leroux, Prog. Lipid Res., 2000, 39, 409 CrossRef CAS.
- M. Keller, R. P. Harbottle, E. Perouzel, M. Colin, I. Shah, A. Rahim, L. Vaysse, A. Bergau, S. Moritz, C. Brahimi-Horn, C. Coutelle and A. D. Miller, ChemBioChem, 2003, 4, 286 CrossRef CAS.
- L. Vaysse, R. Harbottle, B. Bigger, A. Bergau, O. Tolmachov and C. Coutelle, J. Biol. Chem., 2004, 279, 5555 CAS.
- J. L. Young, J. N. Benoit and D. A. Dean, Gene Ther., 2003, 10, 1465 CrossRef CAS.
- K. Kogure, R. Moriguchi, K. Sasaki, M. Ueno, S. Futaki and H. Harashima, J. Controlled Release, 2004, 98, 317 CrossRef CAS.
- R. G. Jenkins, S. E. Herrick, Q. H. Meng, C. Kinnon, G. J. Laurent, R. J. McAnulty and S. L. Hart, Gene Ther., 2000, 7, 393 CrossRef CAS.
- M. Colin, R. P. Harbottle, A. Knight, M. Kornprobst, R. G. Cooper, A. D. Miller, G. Trugnan, J. Capeau, C. Coutelle and M. C. Brahimi-Horn, Gene Ther., 1998, 5, 1488 CrossRef CAS.
- R. G. Cooper, R. P. Harbottle, H. Schneider, C. Coutelle and A. D. Miller, Angew. Chem. Int. Ed., 1999, 38, 1949 CrossRef CAS.
- M. Colin, M. Maurice, G. Trugnan, M. Kornprobst, R. P. Harbottle, A. Knight, R. G. Cooper, A. D. Miller, J. Capeau, C. Coutelle and M. C. Brahimi-Horn, Gene Ther., 2000, 7, 139 CrossRef CAS.
- J. E. Waterhouse, R. P. Harbottle, M. Keller, K. Kostarelos, C. Coutelle, M. R. Jorgensen and A. D. Miller, ChemBioChem, 2005, 6, 1212 CrossRef CAS.
- S. Kawakami, A. Sato, M. Nishikawa, F. Yamashita and M. Hashida, Gene Ther., 2000, 7, 292 CrossRef CAS.
- S. Kawakami, A. Sato, M. Yamada, F. Yamashita and M. Hashida, Stp Pharma. Sci., 2001, 11, 117 Search PubMed.
- S. Simoes, V. Slepushkin, P. Pires, R. Gaspar, M. P. de Lima and N. Duzgunes, Gene Ther., 1999, 6, 1798 CrossRef CAS.
- M. T. G. da Cruz, S. Simoes, P. P. C. Pires, S. Nir and M. C. P. de Lima, Biochim. Biophys. Acta, 2001, 1510, 136 CAS.
- C. T. de Ilarduya and N. Duzgunes, Biochim. Biophys. Acta, 2000, 1463, 333.
- P. H. Tan, W. J. King, D. Chen, H. M. Awad, M. Mackett, R. I. Lechler, D. F. P. Larkin and A. J. T. George, Transplantation, 2001, 71, 552 Search PubMed.
- S. Y. Joo and J. S. Kim, Drug Dev. Ind. Pharm., 2002, 28, 1023 CrossRef CAS.
- K. Yanagihara, H. Cheng and P. W. Cheng, Cancer Gene Ther., 2000, 7, 59 CrossRef CAS.
- N. Joshee, D. R. Bastola and P. W. Cheng, Hum. Gene Ther., 2002, 13, 1991 CrossRef CAS.
- S. Simoes, V. Slepushkin, P. Pires, R. Gaspar, M. C. P. de Lima and N. Duzgunes, Biochim. Biophys. Acta, 2000, 1463, 459 CAS.
- K. Yanagihara and P. W. Cheng, Biochim. Biophys. Acta, 1999, 1472, 25 CrossRef CAS.
- L. Xu, P. Frederik, K. F. Pirollo, W. H. Tang, A. Rait, L. M. Xiang, W. Huang, I. Cruz, Y. Yin and E. H. Chang, Hum. Gene Ther., 2002, 13, 469 CrossRef CAS.
- T. C. de Ilarduya, M. A. Arangoa, M. J. Moreno-Aliaga and N. Duzgunes, Biochim. Biophys. Acta, 2002, 1561, 209.
- M. Seki, J. Iwakawa, H. Cheng and P. W. Cheng, Hum. Gene Ther., 2002, 13, 761 CrossRef CAS.
- J. G. Seol, D. S. Heo, H. K. Kim, J. H. Yoon, B. I. Choi, H. S. Lee, N. K. Kim and C. Y. Kim, In Vivo, 2000, 14, 513 Search PubMed.
- M. Voinea, E. Dragomir, I. Manduteanu and M. Simionescu, Vasc. Pharmacol., 2002, 39, 13 Search PubMed.
- L. Xu, W. H. Tang, C. C. Huang, W. Alexander, L. M. Xiang, K. F. Pirollo, A. Rait and E. H. Chang, Mol. Med., 2001, 7, 723 CAS.
- L. Xu, C. C. Huang, W. Huang, W. H. Tang, A. Rait, Y. Z. Yin, I. Cruz, L. M. Xiang, K. F. Pirollo and E. H. Chang, Mol. Cancer Ther., 2002, 1, 337 Search PubMed.
- W. Yu, K. F. Pirollo, B. Yu, A. Rait, L. Xiang, W. Huang, Q. Zhou, G. Ertem and E. H. Chang, Nucleic Acids Res., 2004, 32, e48 CrossRef.
- W. Yu, K. F. Pirollo, A. Rait, B. Yu, L. M. Xiang, W. Q. Huang, Q. Zhou, G. Ertem and E. H. Chang, Gene Ther., 2004, 11, 1434 CrossRef CAS.
- P. H. Tan, M. Manunta, N. Ardjomand, S. A. Xue, D. F. Larkin, D. O. Haskard, K. M. Taylor and A. J. George, J. Gene Med., 2003, 5, 311 CrossRef CAS.
- L. Mohr, J. I. Schauer, R. H. Boutin, D. Moradpour and J. R. Wands, Hepatology, 1999, 29, 82 CrossRef CAS.
- Y. Kaneda, Mol. Membr. Biol., 1999, 16, 119 CrossRef CAS.
- Y. Yonemitsu, E. W. Alton, K. Komori, T. Yoshizumi, K. Sugimachi and Y. Kaneda, Int. J. Oncol., 1998, 12, 1277 CAS.
- Y. Kaneda, K. Iwai and T. Uchida, Science, 1989, 243, 375 CAS.
- R. Morishita, G. H. Gibbons, Y. Kaneda, T. Ogihara and V. J. Dzau, Gene, 1994, 149, 13 CrossRef CAS.
- M. Aoki, R. Morishita, J. Higaki, A. Moriguchi, I. Kida, S. Hayashi, H. Matsushita, Y. Kaneda and T. Ogihara, Biochem. Biophys. Res. Commun., 1997, 231, 540 CrossRef CAS.
- Y. Saeki, N. Matsumoto, Y. Nakano, M. Mori, K. Awai and Y. Kaneda, Hum. Gene Ther., 1997, 8, 2133 CAS.
- Y. Yonemitsu, Y. Kaneda, A. Muraishi, T. Yoshizumi, K. Sugimachi and K. Sueishi, Gene Ther., 1997, 4, 631 CrossRef CAS.
- M. Nakanishi, H. Mizuguchi, K. Ashihara, T. Senda, A. Eguchi, A. Watabe, T. Nakanishi, M. Kondo, T. Nakagawa, A. Masago, J. Okabe, S. Ueda, T. Mayumi and T. Hayakawa, Mol. Membr. Biol., 1999, 16, 123 CrossRef CAS.
- P. Schoen, A. Chonn, P. R. Cullis, J. Wilschut and P. Scherrer, Gene Ther., 1999, 6, 823 CrossRef CAS.
- E. R. Waelti and R. Gluck, Int. J. Cancer, 1998, 77, 728 CrossRef CAS.
- A. Abe, A. Miyanohara and T. Friedmann, J. Virol., 1998, 72, 6159 CAS.
- D. D. Lasic and D. Papahadjopoulos, Science, 1995, 267, 1275 CAS.
- A. Martin-Herranz, A. Ahmad, H. M. Evans, K. Ewert, U. Schulze and C. R. Safinya, Biophys. J., 2004, 86, 1160 CAS.
- H. L. Chen, Q. H. Hu and W. Q. Liang, Pharmazie, 2004, 59, 131 CAS.
- J. J. Wheeler, L. Palmer, M. Ossanlou, I. MacLachlan, R. W. Graham, Y. P. Zhang, M. J. Hope, P. Scherrer and P. R. Cullis, Gene Ther., 1999, 6, 271 CrossRef CAS.
- Y. P. Zhang, L. Sekirov, E. G. Saravolac, J. J. Wheeler, P. Tardi, K. Clow, E. Leng, R. Sun, P. R. Cullis and P. Scherrer, Gene Ther., 1999, 6, 1438 CrossRef CAS.
- P. Tam, M. Monck, D. Lee, O. Ludkovski, E. C. Leng, K. Clow, H. Stark, P. Scherrer, R. W. Graham and P. R. Cullis, Gene Ther., 2000, 7, 1867 CrossRef CAS.
- E. G. Saravolac, O. Ludkovski, R. Skirrow, M. Ossanlou, Y. P. Zhang, C. Giesbrecht, J. Thompson, S. Thomas, H. Stark, P. R. Cullis and P. Scherrer, J. Drug Target., 2000, 7, 423 Search PubMed.
- L. R. Palmer, T. Chen, A. M. Lam, D. B. Fenske, K. F. Wong, I. MacLachlan and P. R. Cullis, Biochim. Biophys. Acta, 2003, 1611, 204 CAS.
- L. Palmer, L. B. Jeffs, K. P. Y. Chan, K. McClintock, C. Giesbrecht, J. Heyes, A. C. H. Lee and I. MacLachlan, Mol. Ther., 2004, 9, 679.
- A. C. H. Lee, J. C. Evans, J. R. Shaw, L. E. Swann, K. McClintock and I. MacLachlan, Mol. Ther., 2003, 7, 549.
- M. E. Hayes, D. C. Drummond, D. B. Kirpotin, J. W. Park, J. D. Marks, C. C. Benz and K. Hong, Biophys. J., 2004, 86, 36A.
- X. Guo and F. C. Szoka, Jr., Bioconjugate Chem., 2001, 12, 291 CrossRef CAS.
- X. Guo, Z. Huang and F. C. Szoka, Methods Enzymol., 2004, 387, 147 CAS.
- X. Guo, J. A. MacKay and F. C. Szoka, Jr., Biophys. J., 2003, 84, 1784 CAS.
- J. S. Choi, J. A. MacKay and F. C. Szoka, Jr., Bioconjugate Chem., 2003, 14, 420 CrossRef CAS.
- J. Rejman, A. Wagenaar, J. B. Engberts and D. Hoekstra, Biochim. Biophys. Acta, 2004, 1660, 41 CAS.
- J. A. Boomer, H. D. Inerowicz, Z. Y. Zhang, N. Bergstrand, K. Edwards, J. M. Kim and D. H. Thompson, Langmuir, 2003, 19, 6408 CrossRef CAS.
- D. H. Thompson, J. Shin, J. Boomer and J. M. Kim, Methods Enzymol., 2004, 387, 153 CAS.
- B. Pitard, N. Oudrhiri, O. Lambert, E. Vivien, C. Masson, B. Wetzer, M. Hauchecorne, D. Scherman, J. L. Rigaud, J. P. Vigneron, J. M. Lehn and P. Lehn, J. Gene Med., 2001, 3, 478 CrossRef CAS.
- C. Nicolazzi, N. Mignet, N. de la Figuera, M. Cadet, R. T. Ibad, J. Seguin, D. Scherman and M. Bessodes, J. Controlled Release, 2003, 88, 429 CrossRef CAS.
- C. Nicolazzi, N. Mignet, N. Dela Figuera, M. Cadet, R. Ibad, J. Seguin, D. Scherman and M. Bessodes, Mol. Ther., 2004, 9, 475 CrossRef.
- N. Y. Shi and W. M. Pardridge, Proc. Natl. Acad. Sci. USA, 2000, 97, 7567 CrossRef CAS.
- N. Shi, R. J. Boado and W. M. Pardridge, Pharm. Res., 2001, 18, 1091 CrossRef CAS.
- Y. F. Zhang, R. J. Boado and W. M. Pardridge, Pharm. Res., 2003, 20, 1779 CrossRef CAS.
- Y. Zhang, H. Jeong Lee, R. J. Boado and W. M. Pardridge, J. Gene Med., 2002, 4, 183 CrossRef.
- Y. Zhang, C. Zhu and W. M. Pardridge, Mol. Ther., 2002, 6, 67 CrossRef CAS.
- Y. Zhang, R. J. Boado and W. M. Pardridge, J. Gene Med., 2003, 5, 1039 CrossRef CAS.
- H. E. Hofland, C. Masson, S. Iginla, I. Osetinsky, J. A. Reddy, C. P. Leamon, D. Scherman, M. Bessodes and P. Wils, Mol. Ther., 2002, 5, 739 CrossRef CAS.
- J. A. Reddy and P. S. Low, J. Controlled Release, 2000, 64, 27 CrossRef CAS.
- G. Shi, W. Guo, S. M. Stephenson and R. J. Lee, J. Controlled Release, 2002, 80, 309 CrossRef CAS.
- I. Jaaskelainen, S. Peltola, P. Honkakoski, J. Monkkonen and A. Urtti, Eur. J. Pharm. Sci., 2000, 10, 187 CrossRef CAS.
- W. E. Walker and A. D. Miller, Mol. Ther., 2004, 9, 184.
- B. W. Bigger, O. Tolmachov, J. M. Collombet, M. Fragkos, I. Palaszewski and C. Coutelle, J. Biol. Chem., 2001, 276, 23018 CrossRef CAS.
- C. M. Chow, A. Athanassiadou, S. Raguz, L. Psiouri, L. Harland, M. Malik, M. A. Aitken, F. Grosveld and M. Antoniou, Gene Ther., 2002, 9, 327 CrossRef CAS.
- (a) A. F. Kolb, C. J. Coates, J. M. Kaminski, J. B. Summers, A. D. Miller and D. J. Segal, Trends Biotechnol., 2005, 23, 399 CrossRef CAS; (b) C. J. Coates, J. M. Kaminski, J. B. Summers, D. J. Segal, A. D. Miller and A. F. Kolb, Trends Biotechnol., 2005, 23, 407 CrossRef CAS.
- G. J. Schouten, H. G. van Luenen, N. C. Verra, D. Valerio and R. H. Plasterk, Nucleic Acids Res., 1998, 26, 3013 CrossRef CAS.
- S. R. Yant, L. Meuse, W. Chiu, Z. Ivics, Z. Izsvak and M. A. Kay, Nat. Genet., 2000, 25, 35 CrossRef CAS.
- Z. Izsvak, Z. Ivics and R. H. Plasterk, J. Mol. Biol., 2000, 302, 93 CrossRef CAS.
- S. Ortiz-Urda, Q. Lin, S. R. Yant, D. Keene, M. A. Kay and P. A. Khavari, Gene Ther., 2003, 10, 1099 CrossRef CAS.
- T. J. Vigdal, C. D. Kaufman, Z. Izsvak, D. F. Voytas and Z. Ivics, J. Mol. Biol., 2002, 323, 441 CrossRef CAS.
- J. M. Kaminski, M. R. Huber, J. B. Summers and M. B. Ward, FASEB J., 2002, 16, 1242 Search PubMed.
- S. Ortiz-Urda, B. Thyagarajan, D. R. Keene, Q. Lin, M. Fang, M. P. Calos and P. A. Khavari, Nat. Med., 2002, 8, 1166 CrossRef CAS.
- E. C. Olivares, R. P. Hollis, T. W. Chalberg, L. Meuse, M. A. Kay and M. P. Calos, Nat. Biotechnol., 2002, 20, 1124 CrossRef CAS.
- S. Ortiz-Urda, B. Thyagarajan, D. R. Keene, Q. Lin, M. P. Calos and P. A. Khavari, Hum. Gene Ther., 2003, 14, 923 CrossRef CAS.
- A. C. Groth and M. P. Calos, J. Mol. Biol., 2004, 335, 667 CrossRef CAS.
- E. M. Westphal, H. Sierakowska, E. Livanos, R. Kole and J. M. Vos, Hum. Gene Ther., 1998, 9, 1863 CAS.
- C. Magin-Lachmann, G. Kotzamanis, L. D'Aiuto, E. Wagner and C. Huxley, BMC Biotechnol., 2003, 3, 2 CrossRef.
- C. Magin-Lachmann, G. Kotzamanis, L. D'Aiuto, H. Cooke, C. Huxley and E. Wagner, J. Gene Med., 2004, 6, 195 CrossRef CAS.
- S. H. Compton, S. Mecklenbeck, J. E. Mejia, S. L. Hart, M. Rice, R. Cervini, Y. Barrandon, Z. Larin, E. R. Levy, L. Bruckner-Tuderman and A. Hovnanian, Gene Ther., 2000, 7, 1600 CrossRef CAS.
- S. Mecklenbeck, S. H. Compton, J. E. Mejia, R. Cervini, A. Hovnanian, L. Bruckner-Tuderman and Y. Barrandon, Hum. Gene Ther., 2002, 13, 1655 CrossRef CAS.
- D. Huertas, S. Howe, A. McGuigan and C. Huxley, Hum. Mol. Genet., 2000, 9, 617 CrossRef CAS.
- G. Vassaux, A. L. Manson and C. Huxley, Gene Ther., 1997, 4, 618 CrossRef CAS.
- J. M. Vos, Nat. Biotechnol., 1997, 15, 1257 CrossRef CAS.
- S. Stewart, N. MacDonald, E. Perkins, G. DeJong, C. Perez and M. Lindenbaum, Gene Ther., 2002, 9, 719 CrossRef CAS.
- G. de Jong, A. Telenius, S. Vanderbyl, A. Meitz and J. Drayer, Chromosome Res., 2001, 9, 475 CrossRef CAS.
- E. Csonka, I. Cserpan, K. Fodor, G. Hollo, R. Katona, J. Kereso, T. Praznovszky, B. Szakal, A. Telenius, G. deJong, A. Udvardy and G. Hadlaczky, J. Cell Sci., 2000, 113(Pt 18), 3207 Search PubMed.
- J. E. Mejia, A. Willmott, E. Levy, W. C. Earnshaw and Z. Larin, Am. J. Hum. Genet., 2001, 69, 315 CrossRef CAS.
- K. Konopka, N. Duzgunes, J. Rossi and N. S. Lee, J. Drug Target., 1998, 5, 247 Search PubMed.
- D. M. Dykxhoorn, C. D. Novina and P. A. Sharp, Nat. Rev. Mol. Cell Biol., 2003, 4, 457 CrossRef CAS.
- A. Khvorova, A. Reynolds and S. D. Jayasena, Cell, 2003, 115, 209 CrossRef CAS.
- D. S. Schwarz, G. Hutvagner, T. Du, Z. Xu, N. Aronin and P. D. Zamore, Cell, 2003, 115, 199 CrossRef CAS.
- K. Ui-Tei, Y. Naito, F. Takahashi, T. Haraguchi, H. Ohki-Hamazaki, A. Juni, R. Ueda and K. Saigo, Nucleic Acids Res., 2004, 32, 936 CrossRef CAS.
- Y. Naito, T. Yamada, K. Ui-Tei, S. Morishita and K. Saigo, Nucleic Acids Res., 2004, 32, W124 CAS.
- X. Guo and F. C. Szoka, Jr., Acc. Chem. Res., 2003, 36, 335 CrossRef CAS.
- P. R. Dash, M. L. Read, K. D. Fisher, K. A. Howard, M. Wolfert, D. Oupicky, V. Subr, J. Strohalm, K. Ulbrich and L. W. Seymour, J. Biol. Chem., 2000, 275, 3793 CrossRef CAS.
- K. Kostarelos and A. D. Miller in Non-viral Vectors for Gene Therapy, ed. L. Huang, M.-C. Hung and E. Wagner, Elsevier Academic Press, 2005 Search PubMed.
Footnote |
| † Present address: Centre for Drug Delivery Research, The School of Pharmacy, University of London, 29–39 Brunswick Square, London WC1 1AX, UK. Email: kostas.kostarelos@ulsop.ac.uk |
| This journal is © The Royal Society of Chemistry 2005 |