Measuring rates of reaction in supercooled organic particles with implications for atmospheric aerosol†
John D.
Hearn
and
Geoffrey D.
Smith
*
Department of Chemistry, University of Georgia, Athens, GA, 30602-2556, USA. E-mail: gsmith@chem.uga.edu; Fax: +1 (706) 542-9454; Tel: +1 (706) 583-0478
First published on 31st May 2005
Abstract
The kinetics of heterogeneous reactions involving supercooled organic droplets is reported for the first time. Reactions between ozone and internally-mixed sub-micrometre particles containing an unsaturated alkenoic acid, oleic acid, and an n-alkanoic acid, myristic acid, were studied as a simple model for the oxidation of meat-cooking aerosol. The reactions were followed by monitoring the rate of oleic acid loss using an Aerosol CIMS (chemical ionization mass spectrometry) instrument for real-time particle analysis. Evidence of as much as 32 °C supercooling at room temperature was observed depending on particle composition. FTIR spectra of the aerosol also demonstrate features indicative of supercooling. Particles in which crystallization was induced by cooling below room temperature demonstrated decreased reactivity by a factor of 12 compared to supercooled particles of the same composition. This drastic difference in reactivity could have significant implications for the lifetimes of reactive species in ambient aerosol as well as for the accurate source apportionment of particulate matter.
Organic aerosol in the atmosphere consists of complex mixtures containing as many as hundreds of separate species.1 This condensed-phase organic material is abundant throughout the lower atmosphere, and in the US it comprises 20–40% of the fine particulate mass in rural areas and 30–80% in urban locations.2 While in the atmosphere these particles react with trace gas-phase species potentially altering their toxicity, hygroscopicity, ability to act as cloud condensation nuclei and ability to reflect sunlight. However, the fundamental processes determining the rates of this transformation have not been fully identified and characterized. Here, we demonstrate for the first time that mixed organic particles can exist in a non-equilibrium state as supercooled droplets drastically affecting their reactivity. Particles supercooled by up to 32 °C are observed to react with ozone more than an order of magnitude more quickly than crystallized particles of the same composition. These droplets are estimated to remain supercooled in the atmosphere for up to days with significant implications for the lifetimes of reactive aerosol species as well as the accurate source apportionment of particulate matter.
Recently, many different researchers have investigated the oxidation kinetics of organic particles by studying a simple, representative reaction: the reaction of ozone with oleic acid particles or films.3–10 Oleic acid is an unsaturated carboxylic acid and is liquid at room temperature. It is a major component of meat-cooking aerosol, found to account for as much as 21% of the primary fine organic particle emissions in the Los Angeles area.11 Oleic acid originates from the vaporization of fat and composes as much as 23% of the identifiable organics in meat-cooking aerosol.11
Though laboratory measurements of the rate of this reaction are in fairly good agreement with one another,3,4,6,8–10 the rate is orders of magnitude too fast to explain the observed abundance of atmospheric particulate oleic acid.4,11 Ambient particles are not pure but consist of many different species; in particular, the largest components of meat-cooking aerosol other than oleic acid are alkanoic acids such as myristic acid, palmitic acid and stearic acid (solids at room temperature).11 A few researchers have recently begun investigating the effect of this so-called particle “matrix” on reactivity,9,10 indicating that particle phase and morphology may be important factors.
We have taken a step towards understanding how the composition of a particle matrix can influence the reactivity of particular components by studying the kinetics of internally-mixed particles with solid and/or liquid components. The model system used was the reaction of ozone with 700 nm particles containing oleic acid (OA), C18H34O2, and an alkanoic acid, myristic acid (MA), C14H28O2. These mixtures were used as simple representatives of meat-cooking aerosol which includes both of these species as major constituents.11 The particles were generated through homogeneous nucleation of heated vapors (100–150 °C) of the individual components as they cooled to room temperature. This mechanism is convenient in that no solvent is used which could affect the reaction, and it may closely resemble aerosol formation from cooking meat. The particles reacted with approximately 10−4 atm (2.5 × 1015 molecules cm−3) of ozone at 293 K in an aerosol flow tube for a variable reaction time of up to 6 s.12 Rates of reaction were measured by monitoring rates of OA loss using the Aerosol CIMS (chemical ionization mass spectrometry) technique.13
Representative reactive decays for the OA/MA particles are shown in Fig. 1. For each of the room-temperature mixtures (filled symbols) the OA reacts to completion at approximately the same rate as in the pure OA droplets. This finding is surprising since MA is known to be a solid at room temperature14 (see Fig. 2) and is expected to slow the reaction. In fact, Knopf et al. have recently measured the rate of ozone loss on OA/MA films and have seen decreases in the reactivity by as much as a factor of 20 if the MA freezes.10 Likewise, Ziemann has observed a decrease in OA reactivity by up to a factor of 10 in particles containing larger alkanoic acids.9 The apparent discrepancy indicates that the particles used in the present study react as if they are liquid droplets instead of solid–liquid particles. In fact, it turns out that these particles exist as droplets because they can be supercooled, whereas the bulk films and particles of the other studies were solid–liquid mixtures.9,10 Supercooling was not observed in the bulk mixtures of Knopf et al. because it was inhibited by the presence of imperfections at the surface of the flow tube wall. In the work of Ziemann, the composition of the particles and the fact that they were created at room temperature prevented them from being supercooled.
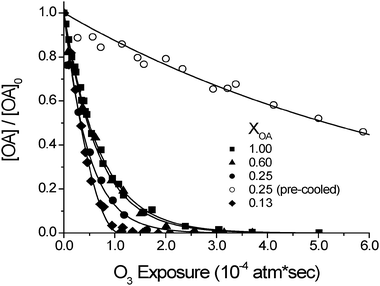 | ||
| Fig. 1 Reaction decays for internally-mixed oleic acid (OA) and myristic acid (MA) particles with ozone. The oleic acid in the supercooled particles (filled symbols) reacts quickly and to completion. The oleic acid in the pre-cooled particles (open circles) reacts more slowly because the myristic acid crystallizes and inhibits ozone solubility and/or diffusion of ozone or oleic acid. Lines are drawn to guide the eye. | ||
 | ||
| Fig. 2 Phase diagram for the OA/MA mixture as calculated from the data of Inoue et al.14,15 The filled squares indicate particle compositions studied in the present work at 293 K. The open square represents particles which were pre-cooled to −2 °C inducing crystallization of the myristic acid before reaction with ozone. The particles with XMA = 0.87 are supercooled by 32 °C, as indicated by the vertical arrow, representing a lower limit on the amount of supercooling attainable. | ||
We tested the possibility that our particles are supercooled by further cooling them to −2 °C, allowing them to warm back up to room temperature and then reacting them with ozone. Fig. 1 shows the reactive decay for these “pre-cooled” particles containing an OA mole fraction of 0.25 (open circles). These particles were observed to react significantly more slowly (by a factor of 12) than particles of the same composition which were not pre-cooled (filled circles). We conclude that pre-cooling the particles induces crystallization of the MA which slows the reaction of OA with ozone. In particular, the solid matrix inhibits the reactivity of OA by reducing solubility of ozone and/or slowing diffusion of either ozone or OA.
Further evidence for supercooling can be seen in FTIR spectra taken of OA/MA particles (Fig. 3). According to the phase diagram (Fig. 2),14,15 at 293 K this mixture should consist of solid MA with a liquid solution containing mostly OA. However, the FTIR spectra demonstrate two features which indicate otherwise. In Fig. 3a the CH2 wag progression (1150–1350 cm−1) is shown. This progression demonstrates well-resolved structure for the OA/MA particles which have been pre-cooled to 0 °C and then warmed up to room temperature (bottom trace), indicative of the solid phase, whereas it is broad and featureless in the supercooled OA/MA spectrum (top trace), characteristic of the liquid phase.16Fig. 3b shows the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretch of the hydrogen-bonded carboxylic acid dimers near 1700 cm−1 for OA/MA particles which have cooled to room temperature (top trace) and which have been pre-cooled (bottom trace). A red shift of 9 cm−1 is evident in the pre-cooled particles. Similar red shifts of 10–11 cm−1 for lauric acid and palmitic acid have been measured by Zhang et al. upon freezing.17 Likewise, the shift observed in our particles indicates that the pre-cooled particles are predominantly solid whereas the particles cooled to room temperature are liquid (supercooled). This shift can be attributed to a weakening in the hydrogen bonding between the acids as the melt solidifies; the solid network imposes a higher degree of order on the tails of the carboxylic acids resulting in a slight pulling apart and weakening of the hydrogen bonds.
O stretch of the hydrogen-bonded carboxylic acid dimers near 1700 cm−1 for OA/MA particles which have cooled to room temperature (top trace) and which have been pre-cooled (bottom trace). A red shift of 9 cm−1 is evident in the pre-cooled particles. Similar red shifts of 10–11 cm−1 for lauric acid and palmitic acid have been measured by Zhang et al. upon freezing.17 Likewise, the shift observed in our particles indicates that the pre-cooled particles are predominantly solid whereas the particles cooled to room temperature are liquid (supercooled). This shift can be attributed to a weakening in the hydrogen bonding between the acids as the melt solidifies; the solid network imposes a higher degree of order on the tails of the carboxylic acids resulting in a slight pulling apart and weakening of the hydrogen bonds.
 | ||
| Fig. 3 FTIR spectra of OA/MA particles with XOA
= 0.32. The top trace is of supercooled liquid droplets and the bottom trace is of “pre-cooled” solid–liquid particles. Two features indicate that the pre-cooled particles contain solid MA: (a) the CH2 wag progression at 1200–1300 cm−1 is much more clearly defined in the solid, (b) the CO stretch near 1700 cm−1 is red shifted by 9 cm−1 in the solid. | ||
The degree of supercooling cannot be measured directly in these experiments, but it can be estimated from the kinetic data. If we assume that the complete reaction of OA indicates that the particles exist as liquid droplets, then we can place a lower limit on the amount of supercooling in these OA/MA particles. At room temperature the particles with the largest enrichment in MA (XMA = 0.87) show the largest supercooling of 32 °C (see Fig. 2). This amount of supercooling is not unprecedented, as water droplets are well known to supercool by as much as 42 °C18 and droplets of n-alkanes have been observed to supercool by ∼10 °C.19 Oleic acid, itself, has even been supercooled by 9 °C on a clean glass surface,6 representing a lower limit on the supercooling that should be attainable in our particles.
Many researchers have studied the supercooling of aqueous droplets and the reactivity of metastable inorganic-water or organic-water droplets,20,21 and supercooled particles are known to influence the catalytic destruction of stratospheric ozone over the poles of the Earth.22 However, the impact of supercooled organic particles on the chemistry of the lower atmosphere has been unexplored. While there has been no direct evidence to date that supercooled organic particles exist in the atmosphere, many ambient particles are created at elevated temperatures (in meat cooking and in combustion, for example) and these may be supercooled. The cooking of meat, in particular, may produce supercooled particles since it typically reaches temperatures of 100–160 °C,11 comparable to the temperatures we employ in the laboratory (100–150 °C) when creating our mixed particles. The OA in these supercooled particles would then react with ambient ozone much more quickly than in crystallized particles in which it could be slowed or trapped. For example, OA in 200 nm solid–liquid particles exposed to a typical ozone concentration of 100 ppb would have a lifetime of 60 min based on the observed rate of reaction in the crystallized OA/MA particles (see Fig. 1). The OA in 200 nm supercooled droplets reacts much more quickly and would have a lifetime of only five minutes. The much longer lifetime of ambient oleic acid, implied by the fact that it is observed in particulate matter,11 may seem to indicate, therefore, that it does not exist in supercooled droplets. However, it is possible that the particles are supercooled for a short amount of time during which the OA reacts quickly before the solid matrix is formed. The temporary existence of supercooled droplets increases the fraction of OA that exists as a mobile liquid free to diffuse and react with ozone. The observed lifetime of OA would then be shorter than expected since it would reflect the reactivity in both the supercooled droplet and the solid–liquid particle that forms. Thus, the lifetime of supercooling may influence the chemical lifetime of reactive species.
Though we cannot measure the rates of nucleation in the supercooled droplets, we can place a lower limit on their lifetimes. Using polydisperse particles with a mean size of 950 nm and XOA = 0.32, we observed no change in reactivity over the course of 30 min, indicating that these particles remain supercooled at room temperature (by 27 °C) for at least this long. We calculate, then, that a 200 nm particle will stay supercooled at room temperature much longer, for at least 54 h, assuming that the nucleation rate is proportional to the droplet volume. This lifetime is on the same timescale as its residence time in the atmosphere indicating that supercooling has the potential to substantially alter the reactivity of ambient organic aerosol. The lower temperatures of the troposphere are expected to significantly shorten the lifetime of supercooling, but the much faster reactivity in the droplets while they are supercooled will substantially shorten the observed lifetime of reactive species. Also, higher nucleation rates may inhibit reactions in larger particles leading to a preferential reactivity of the smaller, supercooled particles with potential impacts on cloud formation and human health.
Ambient particles are much more complicated than the binary mixtures used in the present study, and how many of the particles supercool will depend on many factors including their compositions, the manners in which they are formed, their histories and the phase diagrams for these complex mixtures. Recent work by Marcolli et al.23 has even shown that small amounts of organic species may prevent particles from becoming completely solid and that the equilibrium state of the organic fraction may be entirely liquid. In that case, the particles will not be supercooled, but the effect would be expected to be the same: rates of reaction would be enhanced in the liquid as compared to a solid–liquid particle. The present work, along with the recent work of Ziemann9 and Knopf et al.,10 demonstrate that accurate particulate source apportionment and understanding of the reactive transformation of organic aerosol will require an accurate account of particle morphology and phase (including supercooling). Additional studies to determine the extent to which ambient, or at least more representative, organic particles are supercooled and how morphology influences reactivity would be beneficial.
Acknowledgements
We gratefully acknowledge support provided by the National Science Foundation (ATM-0402226) and the American Chemical Society Petroleum Research Fund. The authors would also like to thank Allan Bertram and Daniel Knopf for helpful discussions.References
- J. J. Schauer and G. R. Cass, Environ. Sci. Technol., 2000, 34, 1821 CrossRef CAS.
- M. C. Jacobson, H. C. Hansson, K. J. Noone and R. J. Charlson, Rev. Geophys., 2000, 38, 267 CrossRef CAS.
- T. Moise and Y. Rudich, J. Phys. Chem. A, 2002, 106, 6469 CrossRef CAS.
- J. W. Morris, P. Davidovits, J. T. Jayne, J. L. Jimenez, Q. Shi, C. E. Kolb, D. R. Worsnop, W. S. Barney and G. Cass, Geophys. Res. Lett., 2002, 29 DOI:10.1029/2002GL014692.
- G. D. Smith, E. Woods, III, C. L. DeForest, T. Baer and R. E. Miller, J. Phys. Chem. A, 2002, 106, 8085 CrossRef CAS.
- T. D. Thornberry and J. P. D. Abbatt, Phys. Chem. Chem. Phys., 2004, 6, 84 RSC.
- Y. Katrib, S. T. Martin, H. -M. Hung, Y. Rudich, H. Zhang, J. G. Slowik, P. Davidovits, J. T. Jayne and D. R. Worsnop, J. Phys. Chem. A, 2004, 108, 6686 CrossRef CAS.
- J. D. Hearn, A. J. Lovett and G. D. Smith, Phys. Chem. Chem. Phys., 2005, 7, 501 RSC.
- P. Ziemann, Faraday Discuss., 2005, 130 10.1039/b417502f.
- D. A. Knopf, L. M. Anthony and A. K. Bertram, J. Phys. Chem. A, 2005 Search PubMed , in press.
- W. F. Rogge, L. M. Hildemann, M. A. Mazurek, G. R. Cass and B. R. T. Simoneit, Environ. Sci. Technol., 1991, 25, 1112 CAS.
- More information can be found in the electronic supplementary information (ESI)†.
- J. D. Hearn and G. D. Smith, Anal. Chem., 2004, 76, 2820 CrossRef CAS.
- T. Inoue, Y. Hisatsugu, R. Ishikawa and M. Suzuki, Chem. Phys. Lipids, 2004, 127, 161 CrossRef CAS.
- T. Inoue, Y. Hisatsugu, R. Yamamoto and M. Suzuki, Chem. Phys. Lipids, 2004, 127, 143 CrossRef CAS.
- R. N. Jones, A. F. McKay and R. G. Sinclair, J. Am. Chem. Soc., 1952, 74, 2575 CrossRef CAS.
- J. J. Zhang, J. L. Zhang, S. M. He, K. Z. Wu and X. D. Liu, Thermochim. Acta, 2001, 369, 157 CrossRef CAS.
- P. G. Debenedetti and H. E. Stanley, Phys. Today, 2003, 56, 40 CrossRef CAS.
- I. Weidinger, J. Klein, P. Stoeckel, H. Baumgaertel and T. Leisner, J. Phys. Chem. B, 2003, 107, 3636 CrossRef CAS.
- J. A. Thornton, C. F. Braban and J. P. D. Abbatt, Phys. Chem. Chem. Phys., 2003, 5, 4593 RSC.
- T. Koop, Z. Phys. Chem., 2004, 218, 1231 CrossRef CAS.
- A. R. Ravishankara and D. R. Hanson, J. Geophys. Res., [Atmos.], 1996, 101, 3885 CAS.
- C. Marcolli, B. P. Luo and T. Peter, J. Phys. Chem. A, 2004, 108, 2216 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Materials and methods. See http://www.rsc.org/suppdata/cp/b5/b506424d/ |
| This journal is © the Owner Societies 2005 |