Charge-state resolved mid-infrared spectroscopy of a gas-phase protein
Jos
Oomens
a,
Nick
Polfer
a,
David T.
Moore
a,
Lex
van der Meer
a,
Alan G.
Marshall
bc,
John R.
Eyler
d,
Gerard
Meijer
e and
Gert
von Helden
*e
aFOM Institute for Plasma Physics Rijnhuizen, Edisonbaan 14, NL-3439 MN, Nieuwegein, The Netherlands
bNational High Magnetic Field Laboratory, 1800 E. Paul Dirac Drive, Tallahassee, FL 32310-4005, USA
cDepartment of Chemistry and Biochemistry, Florida State University, Tallahassee, FL 32306, USA
dDepartment of Chemistry, University of Florida, Gainesville, FL 32611-7200, USA
eFritz-Haber-Institut der Max-Planck-Gesellschaft, Faradayweg 4-6, D-14195, Berlin, Germany. E-mail: helden@fhi-berlin.mpg.de; Fax: +49-30-84135600; Tel: +49-30-84135603
First published on 7th March 2005
Abstract
Infrared spectra of a 104 amino-acid protein in the gas phase as a function of its charge state are presented. The spectra contain clearly resolvable bands in the amide I and II spectral regions, as well as a band at 1483 cm−1, which is not observed in solution phase spectroscopy and is especially prominent for the higher charge states. Compared to solution, the amide I band is blue-shifted and the amide II band red-shifted, as expected for species in an environment with reduced hydrogen bonding. The band positions are suggestive of a mostly α-helical structure of the protein and their widths are comparable to those in solution, suggesting a similar conformational distribution.
Along with NMR spectroscopy and X-ray diffraction, infrared spectroscopy is one of the key techniques to determine the conformational structures of protein molecules.1 In infrared experiments, vibrational modes that are sensitive to the higher-order structure, such as the C
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching modes (amide I) and the N–H bending modes (amide II), are used to obtain a global picture of the protein conformation. These investigations are routinely carried out on proteins in solution and while the solution phase presents the natural environment for the protein, it is of fundamental interest to investigate its structure in the gas phase as well, free of any environmental effects. Indeed, gas-phase infrared studies in the 1400–1800 cm−1 spectral range, where the amide I and II modes are located, have recently been reported for several amino acids as well as for di- and tri-peptides, revealing detailed structural and dynamical information.2,3 However, experimental data in that wavelength range on gas-phase proteins is lacking.
O stretching modes (amide I) and the N–H bending modes (amide II), are used to obtain a global picture of the protein conformation. These investigations are routinely carried out on proteins in solution and while the solution phase presents the natural environment for the protein, it is of fundamental interest to investigate its structure in the gas phase as well, free of any environmental effects. Indeed, gas-phase infrared studies in the 1400–1800 cm−1 spectral range, where the amide I and II modes are located, have recently been reported for several amino acids as well as for di- and tri-peptides, revealing detailed structural and dynamical information.2,3 However, experimental data in that wavelength range on gas-phase proteins is lacking.
The advent of matrix assisted laser desorption ionization (MALDI)4 and electrospray ionization (ESI)5 sources has provided us with tools to bring large molecules into the gas phase and, in combination with mass-spectrometric techniques, has revolutionized protein identification and characterization over the last decades.6,7 Structural investigations in the solvent-free gas phase provide information on the intrinsic intramolecular interactions in the biomolecule and can thus help elucidate the driving forces that influence protein conformation and hence aid in solving the ‘protein-folding problem’.8,9 Unfortunately, although standard mass spectrometric techniques yield valuable mass and also sequence information for the protein, usually no conformational information is obtained.
Gas-phase techniques that have been implemented to study protein conformations include ion mobility,10 H/D exchange11 and protein fragmentation12,13 experiments. Using those techniques, effects of, for example, the solvent (e.g. water),14 counterions (e.g. Na+, K+),15 temperature and Coulombic interactions on the protein conformation can be investigated. Mid-IR spectroscopy is a rather direct probe of the structure, but has so far only been demonstrated for amino acids,16,17 and small peptides2 in the gas phase. IR spectra in the hydrogen stretching region around 3300 cm−1 have recently been obtained for the protein ubiquitin.13 Here, we show the IR spectra for several charge states of the protein bovine heart cytochrome c, which is, to our knowledge, the largest molecule ever investigated with mid-IR spectroscopy in the gas phase.
IR studies on proteins in solution are often severely hampered by absorption of the solvent and the deconvolution of solvent contributions from the spectra can be difficult.1 In addition, while a wide mixture of protonation states, salt adducts, aggregation states and non-covalent complexes may be present in solution, mass spectrometric methods can unambiguously select a single charge state of a particular complex. Studies in the gas phase thus offer the opportunity to observe the intrinsic IR spectrum of a protein over a large wavelength range and it is a priori not clear what to expect when such measurements are performed. The absence of solvent and Coulomb repulsion within the protein might cause band shifts. In addition, the conformational freedom (the flexibility) might be very different from what it is in solution and different conformations might cause band broadenings and overlaps.
Infrared spectra of cytochrome c in the gas phase are obtained by multiple photon dissociation spectroscopy of the protein isolated in a Fourier Transform Ion Cyclotron Resonance (FT-ICR) mass spectrometer, by use of the widely tunable high intensity radiation of the Free Electron Laser FELIX18 in Nieuwegein, The Netherlands. The laboratory constructed FT-ICR mass spectrometer is described elsewhere.19 Briefly, it is equipped with a 4.7 T actively shielded superconducting magnet and ions are injected from an external source into the ICR cell via an octopole rf ion guide. Ions are generated in a commercial ESI source (Z-Spray, Micromass UK Ltd) and are fed into the octopole via a quadrupole bender (ABB Extrel).
At a 5 Hz repetition rate, FELIX delivers 5 μs long “macropulses” of high intensity (∼60 mJ) infrared radiation, which consist of a train of ps-long micropulses spaced 1 ns apart. The wavelength can be tuned continuously within a factor of three when keeping the electron beam energy constant, and overall between 5 and 250 μm (2000–40 cm−1). In our experiments the FWHM bandwidth was typically 0.5% of the central wavelength. The infrared laser beam enters the vacuum chamber through a ZnSe window and is focused (FL = 15 cm) in the center of the open-ended ICR cell using reflective optics only.
Typically, a solution of 30 μM bovine cytochrome c (Sigma Aldrich) in a 69∶29∶2 methanol–water–acetic acid mixture is used. To form the potassiated clusters, KCl is added at a concentration of about 1 mM. Mass isolation is effected by a stored waveform inverse Fourier Transform (SWIFT) ICR excitation pulse.20 The pressure in the ICR chamber is ∼2 × 10−7 Torr with the ESI source on, thereby limiting the mass resolution substantially. Nonetheless, it is sufficient for the purposes of this investigation, namely the observation of the loss of a potassium ion, which leads to a reduction of charge state by 1.
Multiply protonated protein bovine cytochrome c molecules are generated in an ESI source and the charge state envelope is centered around a mass-to-charge ratio (m/z) ∼900 (Fig. 1(A)). From the spacings between the peaks, the charge states can be determined unambiguously. As IR-induced fragmentation of the protonated protein turned out to be unobservable, some KCl was added to the solution to form potassiated cytochrome c ions. These can be observed in the mass spectra shown in Fig. 1 as side peaks toward higher m/z with respect to the main peaks of the fully protonated protein. The spacing is the mass difference (mK − mH ∼38 Da) divided by the charge state. The even charge states are isolated (Fig. 1(B)) and subsequently, the ion cloud is irradiated with ∼40 focused FEL pulses. The lowest energy dissociation pathway appears to be the detachment of K+, which leads to a mass difference of 39 and, moreover, to a change in the charge state of −1. Hence, infrared induced dissociation is detected at the m/z channels corresponding to the odd charge states, as shown in Fig. 1(C) for an infrared wavelength of 6 μm. The magnitude of the fragment peak is then recorded as the wavelength is tuned to generate an infrared spectrum. Spectra of all even charge states can thus be recorded simultaneously in a multiplexed fashion and spectra for the odd charge states can be recorded in the same way by initially isolating odd charge states. The time delay between ion trapping and photon absorption is on the order of seconds. One can thus reasonably assume that ions are thermalized to 300 K. The binding energy of K+ to aromatic or charged (COO−) groups can be estimated to be 1–2 eV. For the detachment of K+, it is therefore required that many IR photons are absorbed. The precise number will not only depend on the K+ binding energy, but also on factors such as the heat capacities of the ions.
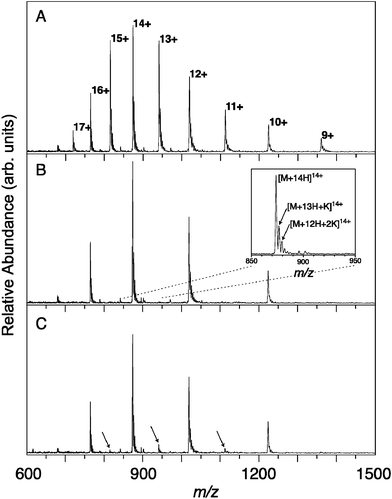 | ||
| Fig. 1 ESI FT-ICR mass spectra of cytochrome c. (A) The full envelope of the different charge states. (B) Mass selective isolation retains the even charge states while ejecting the odd ones from the ICR cell. The inset shows how each peak is composed of the fully protonated peak and partly potassiated peaks. (C) Infrared irradiation at 6 μm, the wavelength of the amide I band, leads to detachment of K+, thereby lowering the charge state by one. The resulting fragment peaks are marked with arrows. | ||
Several independently recorded spectral scans over the 1400–1800 cm−1 range were averaged for both odd and even charge states, as shown in Fig. 2. The spectra for the charge states 12+ to 16+ show three distinct bands in the region of interest at 1483, 1535 and 1660 cm−1. The widths of the bands ranges from 30 to 60 cm−1 and they are therefore substantially broader than the width of the excitation laser in this wavelength range (∼10 cm−1). In solution, the amide I band is usually found between 1625 and 1690 cm−1 and the band at 1660 cm−1 can be assigned to it. The band that we observe at 1535 cm−1 can be assigned to the amide II band, which, in solution, ranges from 1540 to 1550 cm−1. The origin of the 1480 cm−1 band is a priori less clear and will be discussed below.
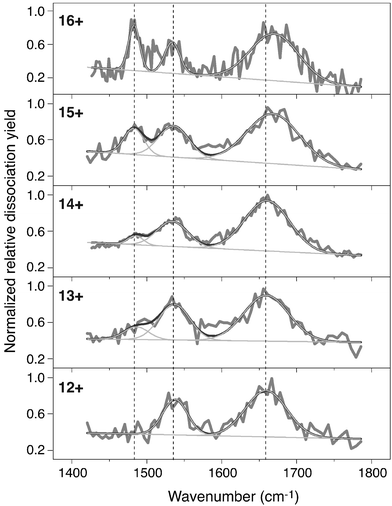 | ||
| Fig. 2 Infrared photodissociation spectra for different charge states of bovine cytochrome c. The black curves represent the average of several independently recorded scans. All spectra are individually normalized. The band at ∼1483 cm−1 clearly grows in with increasing charge state. The band near 1660 cm−1 shows a blue shift as the charge increases. Shown in light gray are fitted Gaussian functions. A total of three Gaussians plus a linear baseline is used. | ||
The amide I mode is considered to be the most diagnostic feature for protein conformation and in solution its position is 1630–1640 cm−1 for antiparallel β-sheets and 1648–1658 cm−1 for α-helices.1 However, calculations for small model systems suggest that there is a blue shift of this vibrational band of between 10 and 80 cm−1 upon going from the solution to the gas phase.21 The amide I band observed here near 1660 cm−1 falls in the range where mainly α-helices are found in solvated proteins. Consistent with this result, about half of the backbone of equine cytochrome c (which has only three point mutations with respect to bovine cytochrome c22) has an α-helix secondary structure23 and a significant fraction of this secondary structure is retained in a denaturing solution24 as well as in the gas-phase charge states under investigation here.11,25 A slight shift of the band of about +10 cm−1 is observed for the 15+ and 16+ states, relative to the lower charge states. It is known from ion mobility studies that the conformation of the gas-phase protein elongates slightly upon increase in charge state over the charge state range studied here.26,27 A higher charge state of the protein would then be expected to display less intramolecular hydrogen bonding, leading to a blue-shift of the CO stretching band.
The amide II band is less frequently used for solution-phase structure assignment. One reason is its overlap with a D2O bending mode, making its determination more difficult. (Obviously, such problems do not arise in gas-phase measurements.) In addition, the band position is less sensitive to conformation, compared to the amide I mode. We observe a band at 1535 cm−1, which is quite close to the solution phase range of 1540–1550 cm−1. In contrast to the amide I mode, the amide II band is known in solution to shift to the red when hydrogen bonding is reduced.28 The absence of solvent and accompanied reduced hydrogen bonding could therefore account for the observed shift.
The band near 1480 cm−1 is harder to assign than the other two, since it has no obvious correspondence with any band observed in solution; yet it is clearly interesting, having by far the strongest dependence on charge state of any of the spectral features. With increasing charge state, it first appears for the 13+ form, and then continues to grow in intensity. At least three explanations for the origin of the 1483 cm−1 band can be given. One possible explanation is that this band is due to the COH in-plane bending (ipb) mode of the COOH side chain groups in the acidic residues Glu and Asp. This could be rationalised if the protein is in the zwitterionic protonation state (where all 23 basic sites are protonated and all 12 acidic sites are de-protonated), resulting in a 11+ charge state. Taking into account the potassium ion and the charge of the heme group (1+),29 the 14+ charge state would thus be the first charge state to have a COO− moiety (of a Glu or Asp) protonated to form COOH, close to the charge state where the 1483 cm−1 band appears. The ipb mode of the COH in a Glu COOH side-chain group in solution is believed to be between 1264 and 1450 cm−1,30 and thus a value of 1483 cm−1 in the gas-phase seems plausible. Upon further protonation, more COOH moieties are formed, thus explaining the linear increase in intensity of this band with increasing charge state. Another conceivable assignment for the 1483 cm−1 mode may be the umbrella mode of the NH3+ moiety, whose absorption is known to be quite intense in the gas phase,17 and for which gas phase17 and matrix isolation studies31 show a band position in close proximity with the 1483 cm−1 band. Cytochrome c contains 18 lysine residues that form NH3+ moieties upon protonation. However, it is as yet unclear how a few oscillators could compete in IR absorbance with the amide I mode, which is carried by more than 100 oscillators.
A third possible explanation is that changes in the secondary structure of the protein give rise to the 1483 cm−1 band. The amide I band, which is the most sensitive probe of secondary structure in solution phase IR spectroscopy, shows a clear blue shift of 10 cm−1 for the highest charge states (15+ and 16+). Such a blue shift is consistent with a possible reduction in the intramolecular hydrogen-bonding in the protein backbone, which could be caused by the greater Coulombic repulsion in the higher charge states. There is an apparently correlated increase in the intensity of the 1483 cm−1 band for these same charge states, suggesting that it is related to the same phenomenon. In addition, ion mobility studies show clear signs of a structural transition, occuring in a charge state range from 10+ to 13+.26,27 At this point, the identity of the vibrational mode at 1483 cm−1 is nonetheless unclear and future deuteration experiments might help to assign this band.
One interesting observation concerns the 50–60 cm−1 amide I band width, which is comparable to the width of this band in the solution-phase spectrum of horse heart cytochrome c.32 The width of the amide I band is determined by the different environments of the individual local oscillators within one protein molecule and also by the distribution of conformers among different protein molecules. Our results thus indicate that this conformational freedom, or so to speak the flexibility, of cytochrome c in the gas phase is not much greater than in the solution phase.
In conclusion, we have presented the first mid-IR spectrum of a gas-phase protein (cytochrome c), which can be measured as a function of charge state and contains resolvable bands that are suggestive of a mostly α-helical structure. The amide I and II bands can be clearly identified. In addition, a third band at lower wavenumbers appears for 13+ and grows in intensity with increasing charge state. The position of the amide I band shows a +10 cm−1 shift for 15+ and 16+, relative to the lower charge states. The positions of the amide I and II bands in the gas phase are blue- and red-shifted with respect to solution respectively, as expected for an environment with reduced hydrogen bonding. The widths of the bands in the gas-phase spectrum are similar to those in solution, indicating a similar conformational distribution.
Clearly, there is a need for a better understanding of the gas-phase spectra of proteins. For cytochrome c, it would be very interesting to observe spectra for the protein in lower charge states, as it is known that electrosprayed proteins may retain their native conformation for lower charge states.26
Acknowledgements
We thank Dr Bateman of Micromass UK Ltd for assistance with the Z-Spray ESI source. This work is part of the research program of FOM, which is financially supported by the Nederlandse Organisatie voor Wetenschappelijk Onderzoek (NWO). Construction and shipping of the FT-ICR instrument were made possible with funding from the National High Field FT-ICR Facility (grant# CHE-9909502) at the National High Magnetic Field Laboratory, Tallahassee, Florida.References
- A. Barth and C. Zscherp, Quart. Rev. Biophys., 2002, 35, 369 CrossRef CAS.
- Special issue “Bioactive molecules in the gas phase”, Phys. Chem. Chem. Phys., 2004, 6, 2543–2890 RSC.
- B. C. Dian, J. R. Clarkson and T. S. Zwier, Science, 2004, 303, 1169 CrossRef CAS.
- M. Karas and F. Hillenkamp, Anal. Chem., 1988, 60, 2299 CrossRef CAS.
- J. B. Fenn, M. Mann, C. K. Meng, S. F. Wong and C. M. Whitehouse, Science, 1989, 246, 64 CrossRef CAS.
- B. T. Chait, R. Wang, R. C. Beavis and S. B. H. Kent, Science, 1993, 262, 89 CrossRef.
- R. Aebersold and M. Mann, Nature, 2003, 422, 198 CrossRef CAS.
- P. Koehl and M. Levitt, Nat. Struct. Biol., 1999, 6, 108 CrossRef CAS.
- D. T. Jones, W. R. Taylort and J. M. Thorton, Nature, 1992, 358, 86 CrossRef CAS.
- M. F. Jarrold, Annu. Rev. Phys. Chem., 2000, 51, 179 CrossRef CAS.
- F. W. McLafferty, Z. Guan, U. Haupts, T. D. Wood and N. L. Kelleher, J. Am. Chem. Soc., 1998, 120, 4732 CrossRef CAS.
- D. W. Price, P. D. Schnier, R. A. Jockusch, E. F. Strittmatter and E. R. Williams, J. Am. Chem. Soc., 1996, 118, 10640 CrossRef CAS.
- H. Oh, K. Breuker, S. K. Sze, Y. Ge, B. K. Carpenter and F. W. McLafferty, Proc. Natl. Acad. Sci. USA, 2002, 99, 15863 CrossRef CAS.
- J. Woenckhaus, Y. Mao and M. F. Jarrold, J. Phys. Chem. B, 1997, 101, 847 CrossRef CAS.
- R. M. Moision and P. B. Armentrout, J. Phys. Chem. A, 2002, 106, 10350 CrossRef CAS.
- J. M. Bakker, L. MacAleese, G. Meijer and G. von Helden, Phys. Rev. Lett., 2003, 91, 203003 CrossRef.
- C. Kapota, J. Lemaire, P. Maitre and G. Ohanessian, J. Am. Chem. Soc., 2004, 126, 1836 CrossRef CAS.
- D. Oepts, A. F. G. van der Meer and P. W. van Amersfoort, Infrared Phys. Technol., 1995, 36, 297 CrossRef CAS.
- (a) D. T. Moore, J. Oomens, L. van der Meer, G. von Helden, G. Meijer, J. Valle, A. G. Marshall and J. R. Eyler, Chem. Phys. Chem., 2004, 5, 740 CrossRef CAS; (b) J. J. Valle, J. R. Eyler, J. Oomens, D. T. Moore, A. F. G. van der Meer, G. von Helden, G. Meijer, C. L. Hendrickson, A. G. Marshall and G. T. Blakney, Rev. Sci. Instrum., 2005, 76, 23103 CrossRef.
- A. G. Marshall, T. C. Wang and T. L. Ricca, J. Am. Chem. Soc., 1985, 107, 7893 CrossRef CAS.
- T. M. Watson and J. D. Hirst, J. Phys. Chem. A, 2003, 107, 6843 CrossRef CAS.
- T. Nakashima, H. Higa, H. Matsubara, A. M. Benson and K. T. Yasunobu, J. Biol. Chem., 1966, 241, 1166 CAS.
- G. W. Bushnell, G. V. Louie and G. D. Brayer, J. Mol. Biol., 1990, 214, 585 CAS.
- Y. Bai, T. R. Sosnick, L. Mayne and S. W. Englander, Science, 1995, 269, 192 CAS.
- Y. Mao, M. A. Ratner and M. F. Jarrold, J. Phys. Chem. B, 1999, 103, 10017 CrossRef CAS.
- M. F. Jarrold, Acc. Chem. Res., 1999, 32, 360 CrossRef CAS.
- S. J. Valentine and D. E. Clemmer, J. Am. Soc. Mass Spectrom., 2002, 13, 506 CrossRef CAS.
- E. Vass, M. Hollósi, F. Besson and R. Buchet, Chem. Rev., 2003, 103, 1917 CrossRef CAS.
- F. He, C. L. Hendrickson and A. G. Marshall, J. Am. Soc. Mass Spectrom., 2000, 11, 120 CrossRef CAS.
- A. Barth, Progress in Biophys. Mol. Biology, 2000, 74, 141 Search PubMed.
- R. Jacob and G. Fischer, J. Phys. Chem. A, 2003, 107, 6136 CrossRef CAS.
- J. O. Speare and T. S. Rush III, Biopolymers, 2003, 72, 193 CrossRef CAS.
| This journal is © the Owner Societies 2005 |