IR–IR double resonance spectroscopy in helium nanodroplets: Photo-induced isomerization
Gary E.
Douberly
,
Jeremy M.
Merritt
and
Roger E.
Miller
*
Department of Chemistry, University of North Carolina, Chapel Hill, NC 27599, USA. E-mail: remiller@unc.edu
First published on 17th December 2004
Abstract
Two IR lasers are used in a pump–probe configuration to observe photo-induced isomerization between the linear and bent isomers of HCN–HF, formed in helium nanodroplets. Vibrational excitation of the C–H and H–F stretching modes of these complexes provides sufficient energy to surmount the barriers between them. The extent of population transfer is found to be different for pumping the two isomers. In the case of linear HCN–HF, the results suggest that the complex undergoes vibrational predissociation, followed by geminate recombination. Excitation of the higher energy bent HF–HCN isomer results in complete population transfer to the linear complex. This isomer specific population transfer provides important clues regarding the associated vibrational dynamics.
Introduction
In recent years there has been rapid progress in the spectroscopic study of molecules solvated in liquid helium. On the one hand, these studies provide information on the properties of the helium solvent and the nature of their interactions with the molecule,1–4 while on the other helium provides an ideal environment for forming new species that are difficult to produce in other ways.5–8 The ability to isolate molecules in a solvent as weak as helium results from the fact that the nanodroplets are wall-less and helium is at least a better solvent than vacuum. Experimental methods now exist for obtaining microwave (MW),9–11 infrared (IR)7,12–16 and visible/UV17,18 spectra of solvated molecules. MW–MW19 and MW–IR10,11,20 double resonance methods have also provided important clues concerning the rotational motion of solvated molecules, while the combination of experimental and theoretical21–25 studies is now providing a quantitative understanding of the rotational dynamics of these systems. For heavy rotors, such as SF6,12 OCS3,13 and cyanoacetylene,10 the effective moment of inertia in helium is typically much larger than in the gas phase, owing to the fact that the helium can follow the molecular motion. For light rotors, including water,26 ammonia,27 HCN14 and acetylene,28 the rotational frequency is so fast that the helium is unable to follow, such that the effective rotational constants are similar to those in the gas phase.The vibrational dynamics of helium solvated molecules that follows IR laser excitation is much less well understood. The ultimate fate of the vibrational excitation is generally known, namely resulting in the evaporation of several hundred helium atoms from the droplet.29 However, the detailed mechanisms associated with these relaxation processes are not well understood. Some indirect evidence for how the molecules are relaxed has come from studies of diatomic HF in helium,30 which has been shown to remain vibrationally excited for the entire flight time of the droplets through the apparatus.31 This is determined by the fact that laser induced excitation of the HF results in an increase in the energy of the droplet beam, as measured by a bolometer detector.32 The implication is that for a diatomic molecule, where there are no intermediate states between the ground and excited vibrational levels, vibrational energy transfer to the helium is very slow. In contrast, polyatomic molecules typically have a whole ladder of vibrational levels, which allows the system to relax in a sequential set of smaller energy steps, corresponding to a cascading of the molecule through at least some of these intermediate levels. To date, however, experiments have not been done which directly probe the transient population in these intermediate levels.
In the present study we report results obtained using an IR pump–IR probe technique which could ultimately be used to probe these intermediate levels. However, since the time resolution of the present experiments is limited by the use of continuous wave lasers, we begin by considering the application of the method to the study of photo-induced isomerization of a hydrogen-bonded complex, due to vibrational excitation. Gas phase complexes are well known to undergo vibrational predissociation when the excited intramolecular vibrational mode has an energy that is greater than the hydrogen bonding energy.33–37 Indeed, much is already known about the associated energy transfer processes that funnel energy from the intramolecular vibrational coordinate into the intermolecular bond. The solvation of a complex in helium introduces a range of other possible dynamical pathways. For example, if the vibrational predissociation lifetime is long with respect to the relaxation rate to the helium, it is possible that intramolecular vibrational excitation will not result in dissociation of the complex, even when the latter channel is energetically open.
The IR–IR double resonance method used here has been discussed previously in a recent paper from our group,29 where cyanoacetylene was vibrationally excited in helium droplets and then probed again after vibrational cooling was complete. In this earlier study, where isomerization of the molecule is not possible, the results clearly show that the effect of the first photon is simply to reduce the size of the droplet, due to helium evaporation. In the present study we apply this method to the hydrogen bonded HCN–HF binary complex, which has two well characterized isomers in helium droplets.38 This system is thus ideally suited to studying photo-induced isomerization, particularly given that the energies of these two isomers are quite different.
Experimental method
Much of the apparatus used in the present experiments has been described in detail previously.39 Helium droplets are formed in this instrument by expanding helium gas through a 5 μm diameter nozzle, operated at 22.5 K with a backing pressure of 60 bar. In all of the studies reported here, the mean helium droplet size corresponds to approximately 3000 helium atoms.40,41 The droplet beam is collimated by a 0.4 mm skimmer located 1 cm from the nozzle. The droplets then pass through a 3.5 cm long pick-up cell42 containing low pressures of HCN and HF that are independently optimized for the pick-up of one molecule of each type.1 The molecules rapidly find one another within the helium droplet and fuse to form a binary complex, which is cooled to the droplet temperature, namely 0.37 K.2 The condensation energy associated with cluster formation is rapidly removed by the helium droplet, resulting in the evaporation of several hundred helium atoms.42 In our previous study38 we found that the linear HCN–HF isomer is formed approximately twice as often as the higher energy bent HF–HCN complex. The relative populations of these two isomers are controlled more by the approach geometries of the two molecules than by their relative energies.7,38,43The seeded droplets pass through two laser excitation regions separated by 8 cm, corresponding to a droplet flight time of 175 μs. Each of these regions contains many laser crossings, resulting from the use of spherical mirror multipass cells.44 As noted above, vibrational excitation of the molecules in the helium droplet eventually leads to vibrational relaxation and the evaporation of several hundred helium atoms.29 The resulting decrease in the number of helium atoms reaching the bolometer32 is detected by amplitude modulating the laser and using phase sensitive detection.
The 175 μs time between the pump and probe lasers is sufficient to ensure that the excited molecules have cooled back to the ground state, prior to their entering the interaction region of the downstream probe laser.29 The probe laser used in the present study is a cw F-center laser (Burleigh FCL-20), operating on crystals No. 2 (KCl : Li) or No. 3 (RbCl : Li), pumped by 3.6 or 1.5 W of red light, respectively, from a krypton ion laser. Details on the tuning and calibration of this laser are given elsewhere.35 The pump laser is a cw periodically poled, lithium niobate optical parametric oscillator (PPLN-OPO) (Linos Photonics).45,46 The PPLN crystal is pumped by a single mode, diode pumped cw YAG laser, yielding a bandwidth of less than 1 MHz. The idler beam has a cw power of approximately 60 mW in the spectral region of interest here, namely 3300 cm−1.
Each multipass cell is equipped with Stark electrodes so that the polar complexes can be oriented using a large DC electric field (∼30 kV cm−1) applied to the laser interaction region.47,48 By aligning the laser polarization direction parallel to the applied DC field, the entire ro-vibrational spectrum can be collapsed into a single (pendular state) transition,49 resulting in a considerable improvement in both the pump and probe signals. The pump–probe results reported here were obtained by amplitude modulating the PPLN-OPO (pump), which is held fixed in resonance with the pendular transition associated with one of the vibrational bands of an isomer of the HCN–HF binary complex. The un-modulated F-center laser (probe) is then tuned through the same or other vibrational bands of the two isomers. Phase sensitive detection gives rise to a constant background signal from the PPLN-OPO laser excitation, on top of which rides the signal from the F-center laser. In this modulation scheme, the deviation from the constant background signal, as a function of the F-center laser frequency, corresponds to the difference in the IR pendular spectrum between the pump laser being on and off. Since population can be transferred either into or out of the probed isomer, the resulting double resonance signals can be either positive or negative, relative to this background signal level, as discussed in detail elsewhere.29
Results
In a recent paper, on the IR spectra of hydrogen cyanide–hydrogen fluoride complexes in helium droplets,38 we reported the first high resolution IR spectra of the bent HF–HCN isomer, as well as helium nanodroplet spectra for the well studied (in the gas phase) HCN–HF linear isomer, the latter corresponding to the global minimum on the potential energy surface. This is yet another example where cluster formation in helium nanodroplets5–8,49,50 results in the formation of highly metastable species. The zero-field and Stark spectra of these two isomers of HCN–HF provide us with the rotational constants and dipole moments needed to understand the pendular spectra of these species. Fig. 1(i) shows four pendular spectra recorded using the F-center laser, with a large DC electric field applied to the corresponding interaction region. These four spectra correspond to the C–H and H–F vibrations of the two isomers. The pendular spectra show only a single peak since in parallel polarization, all of the corresponding transitions occur at the same frequency. | ||
| Fig. 1 Pendular spectra (i) of the C–H and H–F stretching vibrations of the bent and linear isomers of HCN–HF in helium droplets. The probe laser was scanned over each of the transitions associated with the vibrational modes indicated along the top of the figure. The double resonance spectra (ii)–(v) were obtained by fixing the OPO pump to the frequency corresponding to the peak in the pendular spectrum (the pumped vibration is shown to the right of the figure) and then scanning the F-center probe laser through each pendular transition. A negative/positive signal in the double resonance spectrum corresponds to a reduction/increase in the population of the pumped isomer. The source conditions were held fixed for all single and double resonance spectra at a backing pressure of 60 bar and a nozzle temperature of 22.5 K producing a mean droplet size of approximately 3000 helium atoms.40 | ||
It is clear from this series of pendular spectra that the line width associated with the hydrogen bonded vibrational mode of the linear isomer is significantly larger than the corresponding free C–H stretch. (Note that the frequency scale associated with the bonded H–F stretch of the linear isomer is ten times that for the other modes, indicative of the much greater broadening in this band.) This is consistent with what has been observed previously in the gas phase, where the difference is even larger. Indeed, the width of the gas phase ν1 band (H–F stretch) of the linear isomer is 230 times larger than that of the ν2 band (C–H stretch),37 which is rationalized by the fact that the H–F stretch is much more strongly coupled to the intermolecular degrees of freedom, compared with that of the C–H stretch, making the vibrational predissociation rate for the H–F stretch correspondingly larger. In the helium nanodroplet pendular spectra the line width of the H–F stretch is 15 times that of the C–H stretch. The smaller difference is at least in part due to the fact that the broadening associated with the C–H stretch of the linear isomer has a significant contribution from inhomogeneous broadening, due to the droplet size distribution.41 Indeed, the asymmetric line shape of the corresponding pendular spectrum shows a tail to the blue, which is characteristic of broadening due to the distribution of droplet sizes in the beam.29
It is also interesting to note that the line width associated with the ν1 band of the linear isomer is 1.6 times that obtained in the gas phase,37 the implication being that the vibrational relaxation rate (corresponding to vibrational predissociation in the gas phase) is enhanced by the presence of the helium solvent. This effect has been observed previously for HCN dimer,39 where the vibrational relaxation rate in helium was found to be 40 times faster than in the gas phase. This makes qualitative sense given that gas phase vibrational predissociation of small complexes is quite often in the sparse density of states regime. Thus the presence of the elementary excitations of the helium can provide the additional density of states needed to allow for more nearly resonant, and therefore faster, vibrational relaxation.
The two bands of the bent isomer, corresponding to the free H–F stretch and the bonded C–H stretch, both appear to be inhomogeneously broadened, as indicated by the tails to the blue side of the spectra. In this case the FWHM line width of the bonded C–H stretch (335 MHz) is only slightly broader than the free C–H stretch of the linear complex (291 MHz). This small difference is consistent with the fact that the intermolecular bond in the bent isomer is rather weak, making the associated intramolecular coupling also weak and the corresponding lifetime rather long. The free H–F stretch of the bent isomer is particularly narrow, with a FWHM linewidth of only 77 MHz, again reflective of the weak coupling in this complex.
We now turn our attention to the pump–probe experiments that are shown in Fig. 1(ii)–1(v). These spectra were all obtained with a large DC electric field applied to the upstream, pump laser interaction region and fixing the pump laser at the peak of the pendular spectra, corresponding to the modes indicated to the right of the figure. Although several different modulation schemes were investigated, these spectra were obtained by amplitude modulating the pump laser, while the downstream probe laser was scanned through the pendular transitions indicated above. In this modulation scheme the double resonance spectra correspond to the difference in the probe laser spectrum, with and without the pump.
Fig. 1(v)(b) shows a double resonance spectrum resulting from pumping and probing the same C–H stretch of the linear isomer. This spectrum has both a negative “hole” and positive “pile”, which previously have been shown to arise from the fact that the pumped molecules that have returned to the same ground state are now in a smaller droplet, which shifts the spectrum to the blue (the pile),29 depicted schematically in Fig. 2. A fit to this double resonance spectrum consisting of two Lorentzians of opposite sign, separated by 162 MHz, yields line widths for the hole and pile of 231 MHz and 258 MHz, respectively, which are both narrower than the overall line width of the single resonance pendular spectrum.
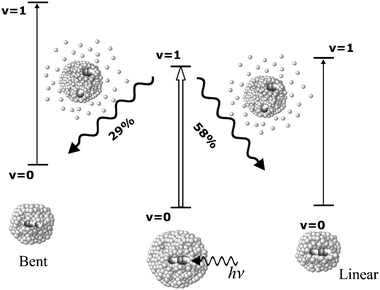 | ||
| Fig. 2 A schematic diagram showing the photo-induced droplet evaporation and cluster isomerization resulting from pumping the linear HCN–HF complex. | ||
In our previous study of the vibrational relaxation of a stable molecule in helium, namely cyanoacetylene, we found that the areas of the hole and pile were approximately the same (within 8%), indicative of the fact that the system has no where else to go but back to the ground state, following vibrational excitation. The small difference was attributed to the fact that the pump process results in a small deflection of the droplet beam, due to helium evaporation, meaning that some of the pumped molecules miss the downstream interaction region and the detector. In contrast, the areas for the linear HCN–HF complex are quite different when pumping and probing the free C–H stretch. Indeed, the signal for the pile only accounts for 58% of the total depletion, indicated by the area of the hole, as is immediately apparent from the spectrum in Fig. 1(v)(b). The same situation is evident in Fig. 1(v)(c) where the H–F stretch of the linear isomer is probed after pumping the corresponding C–H stretch. In this case, the broadening is sufficient to blur the hole and pile into a single peak. The net negative signal clearly indicates that the hole has more intensity than the corresponding pile. So the obvious question is, where is the missing intensity? This question can be answered directly by tuning the probe laser through the C–H and H–F vibrational bands of the bent isomers, as shown in Fig. 1(v)(a) and 1(v)(d), respectively. The positive going signals observed for these two bands clearly show that vibrational excitation of the C–H stretch of the linear isomer results in population transfer to the bent isomer. This photo-induced isomerization process is clearly feasible given the excitation of the C–H stretch of the linear isomer puts the complex approximately 1750 cm−1 above the isomerization barrier.38
A quantitative analysis of the areas for these various processes can provide detailed information on the efficiency of this isomerization process. After correcting for the different transition moments for the various vibrations, using ab initio results,38 as well as for changes in the probe laser intensities for the different spectra, we obtain the results reported in Table 1. These results indicate that 29% of the C–H excited linear complexes find their way into the bent isomer during the relaxation process. When combined with the 58% that cool back into the linear isomer, we have accounted for 87% of the overall population. Excitation of the H–F stretch of the linear isomer gives essentially the same results. At least some of the missing population is indicative of the droplet deflection referred to above.29 The fact that this value is somewhat larger than that observed previously for cyanoacetylene29 could be due to the fact that the droplets used here are on average somewhat smaller, due to the additional evaporation that comes with the formation of the HCN–HF complexes within the droplets. In addition, the use of the approximate ab initio intensities in determining the integrated areas may introduce an error that becomes important when comparing the double resonance spectra for the different isomers. The other possibility is that there is some other minor species that is formed during this photo-excitation process that we have not yet spectroscopically identified. In fact, it is important to note that this approach of pumping and probing the same species can provide us with clues with regards to whether or not a photo-induced process has occurred. Indeed, if the hole is significantly deeper than the pile, we immediately know that there must be some other relaxation channel that is then worth looking for.
| Pump/Probe | L(C–H) | L(H–F) | B(C–H) | B(H–F) |
|---|---|---|---|---|
| L(C–H) | 2.4/1.4 (hole/pile) | 0.9 | 0.7 | 0.7 |
| L(H–F) | … | 1.3 | 0.8 | 0.4 |
| B(C–H) | 1.5 | 2.2 | 2.8 | 2.0 |
| B(H–F) | 1.4 | 3.7 | 1.9 | 3.3 |
One channel we did explore corresponds to the possibility that vibrational predissociation of the complex results in the loss of one of the fragments from the droplet, which would obviously prevent the re-formation of the complex. Indeed, recent studies of CH3I51 and CF3I52 in helium droplets have revealed (at least in this high energy dissociation process) that escape from the droplet is possible. We looked for this channel by tuning the probe laser to the C–H stretch and H–F stretch transitions of the HCN and HF monomers in helium droplets. No double resonance signals were observed in either case, regardless of the isomer pumped. Presumably, the low translational recoil associated with vibrational predissociation53,54 is insufficient to eject the fragments from the droplets. This makes sense given that the solvation energy of a molecule in helium24 is much greater than the translational energies typical of a vibrational predissociation process.53 In contrast, 266 nm photodissociation of CH3I gives rise to 1.3 eV of translational energy,55 suggesting a more ballistic ejection process.
It is interesting to point out that the above results indicate that the linear isomer is formed twice as often as the bent upon excitation of either the C–H or H–F stretch of the linear isomer, namely the same isomer ratio observed in the initial formation of the complexes within the droplets. This result provides a significant clue concerning the mechanism of the population transfer. As shown in Fig. 3, the excitation energy of the linear isomer is 1340 cm−1 above its dissociation limit (given that D0
=
−1970 cm−153), making it reasonable to expect that the complex might undergo vibrational predissociation (similar to that observed in the gas phase) before the energy is transferred to the helium. Subsequent geminate recombination, due to the collision of the fragments with the helium solvent cage, could then result in the re-formation of the complexes with the same isomer ratio as observed in the original pick-up process (see Fig. 2). Indeed, provided that the fragments separate to a large enough distance such that they lose all memory of their relative orientations (the intermolecular interactions become negligible), they will re-condense with the same isomer ratio as obtained from pick-up, assuming they are also internally cold. This seems possible since the diameter of a 3000 helium atom droplet is approximately 6.4 nm ( ), while the interaction energy of the two fragments at 4 nm is approximately 0.4 K (with the dipole moments of the fragments aligned), as determined from the ab initio results. Of course, the results reported here are only suggestive of the mechanism, and we cannot rule out the possibility that isomerization occurs without dissociation and that the 2 : 1 isomer ratio observed for the photo-induced processes is purely a coincidence.
), while the interaction energy of the two fragments at 4 nm is approximately 0.4 K (with the dipole moments of the fragments aligned), as determined from the ab initio results. Of course, the results reported here are only suggestive of the mechanism, and we cannot rule out the possibility that isomerization occurs without dissociation and that the 2 : 1 isomer ratio observed for the photo-induced processes is purely a coincidence.
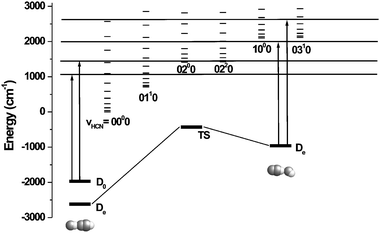 | ||
| Fig. 3 An energy level diagram showing the relative energies of the two HCN–HF isomers and the corresponding transition state, obtained at the MP2/aug-cc-pVTZ level of theory. The arrows indicate the vibrational excitation energy of the C–H and H–F stretching vibrations, while the horizontal bars indicate the available energy. The rotational states of HF are built on top of the vibrational states of HCN, representing the open photo-fragment channels for the binary complex, assuming vibrational excitation is followed by dissociation. | ||
We now turn our attention to the double resonance spectra shown in Fig. 1(ii) and 1(iii), corresponding to pumping the H–F and C–H stretching vibrations of the higher energy bent isomer. The vibrational energy associated with H–F excitation puts the bent complex 3400 cm−1 above the isomerization barrier and 3000 cm−1 above the dissociation limit (see Fig. 3), while the corresponding values for C–H stretch excitation are 2800 cm−1 and 2400 cm−1. These values are considerably larger than those for the linear isomer, which might result in different dynamical processes. It is evident from these double resonance spectra that photo-induced population transfer between the two isomers is again occurring, although the associated dynamics are qualitatively different than those discussed above. The most obvious difference is that only the depletion signal (the “hole”) is observed when pumping and probing the modes of the bent isomer. This is despite the fact that the corresponding double resonance spectra are sharp, suggesting that the depletion signal is not simply the sum of the hole and pile (as is the case for the results in Fig. 1(iv)(c) and 1(v)(c)). The implication is that none of the excited bent complexes make it back into the ground state of the bent complex. Instead, the entire pumped population appears to have been transferred to the linear isomer upon vibrational excitation. This is confirmed by probing the linear isomer, following pumping of the bent isomer, where the corresponding signals are now all positive. The quantitative results are given in Table 1. These isomer specific processes are clearly of considerable interest, even though we do not yet have a complete understanding of them. Indeed, further experimental and theoretical work will be needed to reveal the details associated with these dynamical processes.
Discussion
We have presented here the first observation of photo-induced isomerization of a weakly bound, hydrogen bonded complex embedded in helium droplets. The vibrational excitation energies in the present study are well above the barriers to isomerization between the two isomers of HCN–HF, as well as the dissociation energies of the complexes. The results clearly show that the helium solvent does not quench the excited molecules fast enough to prevent isomerization. While the dynamical processes appear to be mode independent, there is a strong dependence upon which isomer is initially excited. Indeed, excitation of either the H–F or C–H stretches of the linear isomer results in the same ratio of isomer populations that is obtained from the initial pick-up process. This at least suggests that complex dissociation is occurring before the helium can fully quench the system, followed by geminate recombination of the cooled fragments. In contrast, pumping the C–H and H–F stretches of the bent isomer results in complete population transfer to the lower energy, linear isomer. As shown in Fig. 3, the energy difference between the two isomers is quite large, making the energetics associated with isomerization in the two directions quite different. The available energy is considerably larger when pumping the bent isomers, which could very well be the determining factor in controlling the dynamics. Indeed, the higher density of states in this region could result in faster intramolecular vibrational redistribution (IVR), compared to that associated with pumping the linear isomer. Higher IVR rates would compete with vibrational predissociation, effectively heating the molecule internally, allowing it to equilibrate into the lower energy isomer structure. IVR would also improve the coupling of the intramolecular vibrational energy to the helium solvent (since the lower frequency vibrations associated with the corresponding dark states56,57 are more easily quenched by the helium28).It is interesting to note that final state distributions of the photo-fragments resulting from C–H stretch and H–F stretch excitation have been measured for the linear complex in the gas phase.53 In both cases it is found that the majority of the available energy appears as HF rotation (i.e. V-R transfer), with the HCN fragment produced in the ground vibrational state. This is despite the fact that the bending excited state of HCN is energetically accessible. If the early time dynamics of the linear isomer are similar in liquid helium, the rotationally excited HF fragment will likely be quenched rapidly by the helium. Indeed, in an earlier study of the HF monomer in helium,30 we found that the J = 1 level was quenched in approximately 12 ps. Thus the production of ground state HCN and rotationally excited HF, which is rapidly quenched (assuming the higher HF rotational states are similarly quenched by the helium), could then give rise to geminate recombination of the cold molecules, producing the same isomer ratio as resulting from pick-up. Alternatively, if the HCN were produced in a vibrationally excited state, the slow vibrational relaxation of this molecule might mean that recombination occurred before the molecule was fully relaxed. In this case, there would be no reason to think that the isomer ratio would be the same as that obtained by the pick-up of ground state molecules.
As noted above, and apparent from Fig. 3, the available energy upon excitation of the bent isomer is much higher than in the case of the linear complex. In the above scenario we propose that faster IVR rates effectively heat the molecule internally, allowing it to rearrange and cool at a slow enough rate so that complete population transfer results. Alternatively, the complex could still vibrationally predissociate, but due to the higher excess energy, result in the population of vibrationally excited states of the HCN fragment. If geminate recombination occurs more rapidly than vibrational relaxation of the HCN fragment (which seems likely given that the typical vibrational relaxation lifetimes in helium are on the nanosecond time scale39), the result would still be the formation of a hot cluster, which could then go on to isomerize into the linear complex. The mechanisms proposed here provide a reasonable explanation for the observed isomer ratios, resulting from pumping the two isomers of HCN–HF. Nevertheless, further experimental and theoretical work will be needed to test these ideas and to glean deeper insights into these interesting phenomena. For example, time resolved pump–probe experiments could be used to observe the transient monomers that result from vibrational predissociation, if it is indeed occurring. Alternately, the transient development of hot bands in the spectrum of the complex could provide insights into IVR mechanisms that might be important.
Summary
IR–IR double resonance spectroscopy has been used as a tool to study the vibrational dynamics of the hydrogen bonded HCN–HF isomers in helium nanodroplets. Following the absorption of the first IR photon, population transfer is observed from the excited complex to a second stable isomer. Pumping the lower energy linear isomer results in the production of both isomers upon cooling by the helium. In this case the isomer ratio is essentially the same as that obtained following the initial pick-up and complexation in the helium droplets. We tentatively explain these results in terms of vibrational predissociation of the linear complex, followed by geminate recombination due to interaction with the solvent cage. The results also suggest that the fragments are cooled by the solvent on a time scale that is fast with respect to geminate recombination.In contrast, vibrational excitation of the metastable, bent isomer results in complete population transfer to the linear complex. The implication seems to be that the complex either does not dissociate, but undergoes IVR to produce a hot cluster, which slowly cools into the more stable linear isomer, or dissociates to produce vibrationally excited fragments that do not cool before recombination occurs, again producing a hot complex which can then isomerize.
The IR–IR double resonance method presented here could also be used to study the photo-initiation of a reaction from a metastable entrance channel complex.58 In this case, the spectral signature of the reaction would be the appearance of a product complex, with the hole associated with the reactant complex having more intensity than the pile. In systems where the hole and the pile in the double resonance spectrum have approximately the same intensity, it would be immediately obvious that reaction has not occurred. Current work in our laboratory is underway to apply this technique to the study of chemical reactions occurring within the superfluid helium solvent, as well as the application of the method to the study of photo-induced isomerization in biomolecule–water complexes.59,60
Acknowledgements
This work was supported by the National Science Foundation (Grant No. CHE-99-87740) and AFOSR. The PPLN-OPO laser used in this research was acquired with funds from DURIP. G. D. would like to acknowledge the Graduate School at UNC-CH for a Royster Fellowship.References
- M. Hartmann, R. E. Miller, J. P. Toennies and A. F. Vilesov, Science, 1996, 272, 1631 CrossRef CAS.
- M. Hartmann, R. E. Miller, J. P. Toennies and A. F. Vilesov, Phys. Rev. Lett., 1995, 75, 1566 CrossRef CAS.
- S. Grebenev, J. P. Toennies and A. F. Vilesov, Science, 1998, 279, 2083 CrossRef CAS.
- M. Hartmann, F. Mielke, J. P. Toennies, A. F. Vilesov and G. Benedek, Phys. Rev. Lett., 1996, 76, 4560 CrossRef CAS.
- K. Nauta and R. E. Miller, Science, 2000, 287, 293 CrossRef CAS.
- K. Nauta, D. T. Moore, P. L. Stiles and R. E. Miller, Science, 2001, 292, 481 CrossRef CAS.
- K. Nauta and R. E. Miller, Science, 1999, 283, 1895 CrossRef CAS.
- G. E. Douberly and R. E. Miller, J. Phys. Chem. B, 2003, 107, 4500 CrossRef CAS.
- A. Conjusteau, C. Callegari, I. Reinhard, K. K. Lehmann and G. Scoles, J. Chem. Phys., 2000, 113, 4840 CrossRef CAS.
- C. Callegari, I. Reinhard, K. K. Lehmann, G. Scoles, K. Nauta and R. E. Miller, J. Chem. Phys., 2000, 113, 4636 CrossRef CAS.
- S. Grebenev, M. Havenith, F. Madeja, J. P. Toennies and A. F. Vilesov, J. Chem. Phys., 2000, 113, 9060 CrossRef CAS.
- M. Meotner, L. W. Sieck, S. Scheiner and X. F. Duan, J. Am. Chem. Soc., 1994, 116, 7848 CrossRef CAS.
- S. Grebenev, M. Hartmann, M. Havenith, B. Sartakov, J. P. Toennies and A. F. Vilesov, J. Chem. Phys., 2000, 112, 4485 CrossRef CAS.
- K. Nauta and R. E. Miller, Phys. Rev. Lett., 1999, 82, 4480 CrossRef CAS.
- C. Callegari, A. Conjusteau, I. Reinhard, K. K. Lehmann and G. Scoles, J. Chem. Phys., 2000, 113, 10535 CrossRef CAS.
- C. Callegari, K. K. Lehmann, R. Schmied and G. Scoles, J. Chem. Phys., 2001, 115, 10090 CrossRef CAS.
- J. Reho, J. Higgins, C. Callegari, K. K. Lehmann and G. Scoles, J. Chem. Phys., 2000, 113, 9686 CrossRef CAS.
- A. Slenczka, B. Dick, M. Hartmann and J. P. Toennies, J. Chem. Phys., 2001, 115, 10199 CrossRef CAS.
- I. Reinhard, C. Callegari, A. Conjusteau, K. K. Lehmann and G. Scoles, Phys. Rev. Lett., 1999, 82, 5036 CrossRef CAS.
- M. Kunze, P. R. L. Markwick, N. Portner, J. Reuss and M. Havenith, J. Chem. Phys., 2002, 116, 7473 CrossRef CAS.
- R. N. Barnett and K. B. Whaley, J. Chem. Phys., 1993, 99, 9730 CrossRef CAS.
- D. Blume, M. Lewerenz and K. B. Whaley, J. Chem. Phys., 1997, 107, 9067 CrossRef CAS.
- Y. Kwon, D. M. Ceperley and K. B. Whaley, J. Chem. Phys., 1996, 104, 2341 CrossRef CAS.
- K. K. Lehmann, Mol. Phys., 1999, 97, 645 CrossRef CAS.
- K. K. Lehmann and C. Callegari, J. Chem. Phys., 2002, 117, 1595 CrossRef CAS.
- R. Frochtenicht, M. Kaloudis, M. Koch and F. Huisken, J. Chem. Phys., 1996, 105, 6128 CrossRef.
- M. Behrens, U. Buck, R. Frochtenicht, M. Hartmann, F. Huisken and F. Rohmund, J. Chem. Phys., 1998, 109, 5914 CrossRef CAS.
- K. Nauta and R. E. Miller, J. Chem. Phys., 2001, 115, 8384 CrossRef CAS.
- J. M. Merritt, G. E. Douberly and R. E. Miller, J. Chem. Phys., 2004, 121, 1309 CrossRef CAS.
- K. Nauta and R. E. Miller, J. Chem. Phys., 2000, 113, 9466 CrossRef CAS.
- C. M. Lindsay, W. K. Lewis and R. E. Miller, J. Chem. Phys., 2004, 121, 6095 CrossRef CAS.
- T. E. Gough, R. E. Miller and G. Scoles, Appl. Phys. Lett., 1977, 30, 338 CrossRef CAS.
- R. E. Miller, Science, 1988, 240, 447 CrossRef CAS.
- L. Oudejans, D. T. Moore and R. E. Miller, J. Chem. Phys., 1999, 110, 209 CrossRef CAS.
- Z. S. Huang, K. W. Jucks and R. E. Miller, J. Chem. Phys., 1986, 85, 3338 CrossRef CAS.
- N. Halberstadt, Ph. Bréchignac, J. A. Beswick and M. Shapiro, J. Chem. Phys., 1986, 84, 170 CrossRef CAS.
- D. C. Dayton and R. E. Miller, Chem. Phys. Lett., 1988, 143, 181 CrossRef CAS.
- G. E. Douberly and R. E. Miller, J. Chem. Phys., 2005, 122 Search PubMed , (in press).
- K. Nauta and R. E. Miller, J. Chem. Phys., 1999, 111, 3426 CrossRef CAS.
- E. L. Knuth, B. Schilling and J. P. Toennies, in 19th International Symposium on Rarefied Gas Dynamics, University of Oxford, 1994, Oxford University Press, Oxford, 1995, Vol. 19, pp. 270–276 Search PubMed.
- M. Lewerenz, B. Schilling and J. P. Toennies, Chem. Phys. Lett., 1993, 206, 381 CrossRef CAS.
- M. Lewerenz, B. Schilling and J. P. Toennies, J. Chem. Phys., 1995, 102, 8191 CrossRef CAS.
- K. Nauta and R. E. Miller, Chem. Phys. Lett., 2001, 346, 129 CrossRef CAS.
- D. Kaur, A. M. De Souza, J. Wanna, S. A. Hammad, L. Mercorelli and D. S. Perry, Appl. Optics, 1990, 29, 119 CrossRef CAS.
- K. Schneider, P. Kramper, S. Schiller and J. Mlynek, Optics. Lett., 2002, 22, 1293 Search PubMed.
- K. Schneider, P. Kramper, O. Mor, S. Schiller and J. Mlynek, OSA Trends in Optics and Photonics Series, 1998, 19(Advanced Solid State Lasers), 256 Search PubMed.
- J. M. Rost, J. C. Griffin, B. Friedrich and D. R. Herschbach, Phys. Rev. Lett., 1992, 68, 1299 CrossRef CAS.
- P. A. Block, E. J. Bohac and R. E. Miller, Phys. Rev. Lett., 1992, 68, 1303 CrossRef CAS.
- K. Nauta, D. T. Moore and R. E. Miller, Faraday Discuss., 1999, 113, 261 RSC.
- F. Dong and R. E. Miller, J. Phys. Chem. A, 2004, 108, 2181 CrossRef CAS.
- M. Drabbels and A. Braun, The 228th American Chemical Society National Meeting, Philadelphia, PA, August 25, 2004 Search PubMed.
- A. Braun and M. Drabbels, Phys. Rev. Lett., 2005 Search PubMed , in press.
- L. Oudejans and R. E. Miller, Chem. Phys., 1998, 239, 345 CrossRef CAS.
- G. E. Ewing, J. Chem. Phys., 1979, 71, 3143 CrossRef CAS.
- Y. Tanaka, M. Kawasaki and Y. Matsumi, Bull. Chem. Soc. Jpn., 1998, 71, 2539 CAS.
- D. J. Nesbitt and R. W. Field, J. Phys. Chem., 1996, 100, 12735 CrossRef CAS.
- J. Keske and B. H. Pate, Annu. Rev. Phys. Chem., 2000, 51, 323 CrossRef CAS.
- D. L. Yang, T. Yu, M. C. Lin and C. F. Melius, J. Chem. Phys., 1992, 97, 222 CrossRef CAS.
- T. S. Zwier, J. Phys. Chem. A, 2001, 105, 8827 CrossRef CAS.
- M. Y. Choi and R. E. Miller, in preparation.
| This journal is © the Owner Societies 2005 |