Building co-crystals with molecular sense and supramolecular sensibility
Christer B. Aakeröy and Debra J. Salmon
Department of Chemistry, Kansas State University, Manhattan, Kansas 66506, USA. E-mail: aakeroy@ksu.edu
First published on 4th July 2005
Abstract
Molecular recognition is typically associated with molecules in solution, but such events are also responsible for organizing molecules in the solid state. Translating principles of molecular recognition to solid-state assembly of heteromeric molecular solids is of key importance to the development of versatile, reliable and practical supramolecular synthesis. In this article we provide an overview of some modular and transferable strategies for the synthesis of binary and ternary supermolecules and co-crystals based upon a hierarchy of intermolecular interactions, notably hydrogen bonds.
 Christer B. Aakeröy | Christer Aakeröy was born in Sweden but thanks to immigrant parents he was lucky enough to acquire a Norwegian citizenship. After a few unglamorous yet invaluable years working in meat-packing factories, as a substitute teacher, and travelling around Europe, he eventually went to University and obtained an M.Sc. in Chemistry (minors in Mathematics, Biology, and Pedagogy) from Uppsala University. After participating in a student exchange program he was unexpectedly offered a Scholarship from BP to carry out research with the ultimate goal of making artificial bones and teeth. Despite the fact that not a single molar was ever made, he gained a Ph.D. in Inorganic Chemistry from the University of Sussex with Ken Seddon. In 1992, Aakeröy received a Fellowship from the Swedish National Academy of Science that allowed him to spend some time at the University of Minnesota in the laboratories of Peggy Etter and Jan Almlöf. He subsequently landed a position as a Lecturer in Inorganic Chemistry at the Queen's University of Belfast. In 1996 he accepted a position as an Assistant Professor at Kansas State University where he was promoted to Associate Professor in 2001. |
 Debra J. Salmon | Debra J. Salmon earned a B.S. degree in Chemistry from Kansas State University in 2005. She has worked with Dr Christer Aakeröy since October 2002, in which time she has gained a deep appreciation for the art of growing co-crystals. She will enter graduate school at the University of Arizona in Fall 2005 to pursue a Ph.D. degree in Chemistry. |
Introduction
What is the most likely outcome when a homogeneous solution containing two different molecular solutes is allowed to evaporate to dryness? Unless a chemical reaction driven by the formation of covalent bonds takes place between the two solutes, one would, as a rule, expect the appearance of two separate molecular solids. This is a manifestation of the inherent structural selfishness of molecules,1 and it is relied upon every time recrystallization is employed as a method of purification. Recrystallization processes represent essential steps during covalent synthetic procedures and are performed on a daily basis in every synthetic laboratory around the world. In the supramolecular laboratory, however, the very same process provides the supramolecular chemist with an opportunity to move in a completely different direction—a co-crystallization is a deliberate attempt at bringing together different molecular species within one periodic crystalline lattice without making or breaking covalent bonds. Recrystallization and co-crystallization processes are, in essence, only distinguishable by their intents. The goal of the former is a homomeric product, whereas the latter procedure strives for a heteromeric product: success for the former means failure for the latter. In general, the odds are stacked firmly in favor of a homomeric product, Scheme 1, so how do we go about developing reliable, effective, and versatile synthetic methods for the directed assembly of heteromeric co-crystals? This article will attempt to provide some practical suggestions by outlining supramolecular synthetic strategies based upon modular hydrogen-bond driven approaches to the design and synthesis of binary and ternary supermolecules and co-crystals. | ||
| Scheme 1 Recrystallization (homomeric) or co-crystallization (heteromeric)? | ||
Covalent vs. non-covalent synthesis
Covalent synthesis has become an enormously powerful discipline2 because organic chemists have been able to establish reproducible links between molecular structure, reactivity, and reaction pathways through systematic studies of innumerable organic reactions. The explicit correspondence between chemical functional groups and their reactivity has provided a foundation for highly efficient construction of new molecules of enormous variety and complexity. The arrival and on-going development of synthetic organic chemistry arguably represent one of the most important scientific stories of the 20th century. The history of organic synthetic chemistry has produced reliable methods for chemical transformations, and names like Grignard, Wittig and Suzuki are prominent members of the collective that the experienced synthetic chemist has labeled “Named reactions”. Each individual reaction can often be described in a concise yet comprehensive manner—“…this is the transformation of A into B, by treatment with X and Y”. These recipes, which often involve specific catalysts and reagents that facilitate covalent coupling reactions between two different molecular fragments, play a crucial role in every aspect of synthetic chemistry.In contrast, supramolecular synthesis3 has not reached anywhere near the same level of sophistication, and despite much progress4,5 we do not yet have access to a ‘dictionary’ that allows us to translate from molecular structure to supramolecular assembly. We have so far only converted a fraction of our considerable understanding of solution-based molecular recognition phenomena into practical solid-state targeted crystal engineering. Specifically, there is much to be done when it comes to the preparation of molecular co-crystals, a synthesis that requires the assembly and spatial organization of different molecular building blocks within the same periodic crystalline lattice. However, there is no doubt that if we could employ our extensive knowledge of molecular structure/function and apply it to the directed assembly of supramolecular species, then we will have taken a crucial step towards new materials that are faster, cheaper, smarter, or more efficient than current alternatives. In addition, we would gain new insight that is directly applicable to molecular-recognition driven biological catalysis and reactivity.
What is a co-crystal?
Crystal engineering is, by definition, a highly interdisciplinary area, which goes some ways towards explaining why unambiguous definitions and systematic nomenclatures have yet to be developed, let alone fully embraced, within this field. The term co-crystal is certainly not well-defined, and in the current literature we encounter terms like molecular complexes, co-crystals, molecular adducts, molecular salts, clathrates, inclusion compounds, etc., that often are meant to describe one and the same family of chemical compounds. The purpose of this article is not to propose new definitions or to weigh in on the current semantic/semiotic debate,6 but it will be necessary to delineate the scientific realm of this Highlight. This article will then adhere (reasonably strictly) to the following rules/definitions:1. Only compounds constructed from discrete neutral molecular species will be considered as co-crystals. Consequently, all solids containing ions, including complex transition-metal ions, are excluded.7
2. Only co-crystals made from reactants that are solids at ambient conditions will be included.8 Therefore all hydrates and other solvates are excluded which, in principle, eliminates compounds that are typically classified as clathrates or inclusion compounds (where the guest is a solvent or a gas molecule).
3. A co-crystal is a structurally homogeneous crystalline material that contains two or more neutral building blocks that are present in definite stoichiometric amounts.
At this point we are essentially left with two families of compounds: binary donor–acceptor complexes and hydrogen-bonded co-crystals. This article will focus heavily upon representatives from the latter category.9
Examples of binary co-crystals
There are, strictly speaking, no “discrete” aggregates within a solid-state framework but it is nevertheless possible to classify an assembly as being 0-D, 1-D, 2-D or 3-D depending upon the type of intermolecular interactions that are present within and between collections of certain molecules.Some examples of 0-D assemblies, Scheme 2, in co-crystals include heteromeric carboxylic acid : carboxylic acid dimers (1 : 1),10 pyridine : carboxylic acid dimers (1 : 1),11 2-aminopyrimidine : carboxylic acid trimers (1 : 2),12 pyridine : bis(hydroxymethyl)biphenyl trimers (2 : 1),13 bipyridine : carboxylic acid trimers (1 : 2),14 2-aminopyrimidine : carboxylic acid tetramers (2 : 2),12,15 bipyridine : resorcinol tetramers (2 : 2),16 3,5-dinitrobenzoic acid : nicotinic acid tetramers (2 : 2),17 isonicotinamide : carboxylic acid tetramers (2 : 2),18 2-pyridone : carboxylic acid pentamers (4 : 1),19 melamine : thymine tetramers (1 : 3),20 melamine : barbital hexamers (3 : 3),21 and tripenylphosphine oxide with a variety of hydrogen-bond donors (1 : 1).22,23
 | ||
| Scheme 2 Four 0-D motifs found in co-crystals created by heteromeric hydrogen-bond interactions. | ||
More complex, multicomponent, co-crystals containing 0-dimensional motifs also include several elegant examples of capsules held together by hydrogen bonds,24Fig. 1.
 | ||
| Fig. 1 Binary co-crystal containing a capsule held together by multiple carboxylic acid–aminopyrimidine interactions.25 | ||
Examples of co-crystals containing chains, ribbons, and other infinite 1-D motifs, Scheme 3, include: bipyridine : dihydroxybenzene,26 melamine : cyanuric acid,27 bipyridine : (fluorinated)dibromobenzene,28 bipyridine : (diiodobenzene, tetraiodoethylene or diiodine),29 2-aminopyridine : dicarboxylic acid,30 triaminopyrimidine : barbituric acid,31 2-aminopyrimidine : dicarboxylic acid,32 1,2,3-trihydroxybenzene : hexamethylenetetramine,33 diols : diamines34, bis-benzimidazole : dicarboxylic acid,35 and 2-amino-5-nitropyrimidine : 2-amino-3-nitropyridine.36
 | ||
| Scheme 3 Four 1-D motifs generated by heteromeric hydrogen-bond interactions. | ||
Many infinite 2-D assemblies have also been constructed including (but not limited to): piperazine : carboxylic acid,37 trithiocyanuric : bipyridine,38 pyridyloxamide : dicarboxylic acid,39 picolylaminocyclohexenone : dicarboxylic acid,40 triazine : uracil,41 tris-(4-pyridyl)triazine : trimesic acid,42 bipyridine : ureylene dicarboxylic acid,43 and isonicotinamide : dicarboxylic acid,18,44Fig. 2.
 | ||
| Fig. 2 A central hexameric motif in the hydrogen-bonded 2-D network in a diaminotriazine : uracil co-crystal.41 | ||
Finally, some examples of 3-D motifs in binary co-crystals include tetrabromoadamantane : hexamethylenetetramine,45 carbamazepine : tetracarboxylic acid-adamantane46 1,4-di-iodotetrafluorobenzene : hexamethylenetetramine,29 and iodoform : hexamethylenetetramine,47Fig. 3.
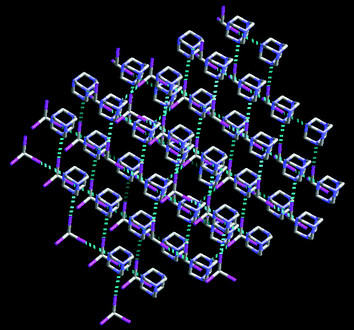 | ||
| Fig. 3 The 3-D network in an iodoform : hexamethylenetetramine co-crystal.47 | ||
From pattern recognition to synthons and practical crystal engineering
The whole notion of structural motifs and patterns within a solid framework is closely linked to the idea that some intermolecular interactions are more important than others.48 The challenge of recognizing and classifying motifs generated by multiple intermolecular interactions was addressed by Etter et al,49 using a system based upon graph-set notation that allow structural motifs to be described in a consistent and ‘user-friendly’ way. The graph-set approach uses four principal motifs: chains (C), dimers (D), rings (R), and intramolecular hydrogen bonds (S), as descriptors of hydrogen-bonded molecular solids. Although this notation may not provide an unambiguous assignment of every structural arrangement, it is flexible enough to facilitate a systematic description of a wide range of structures. An indication of the impact that this nomenclature has had is provided by the fact that two of the early graph-set publications49,50 have together received more than 1,100 citations (SciFinder Scholar, American Chemical Society 2005). An explanation for this success can undoubtedly be found in the appealing simplicity of this approach, and descriptors such as R24(8) (an eight-membered ring with four hydrogen-bond donors and two hydrogen-bond acceptors) and R22(8) (e.g. the carboxylic head-to-head dimer), have become widely recognizable.In the context of structural motifs and pattern preferences, an intriguing and very helpful analogy between covalent and supramolecular synthesis has been captured with the term ‘supramolecular synthon’,51 a robust, transferable connector that can be used for linking molecules together using non-covalent interactions. Synthons describe recognition events that take place when molecules assemble into supermolecules and offer an important illustration of the conceptual similarities between retrosynthetic organic synthesis and supramolecular assembly.52
One of the attractions of supramolecular chemistry is the extraordinary potential for synthesis of new materials that can be achieved much more rapidly and more effectively than with conventional covalent means. For supramolecular synthesis to advance, it is obviously important to characterize, classify, and analyze structural patterns, space group frequencies, and symmetry operators.53 However, at the same time we also need to bring together this information with the explicit aim of improving and developing supramolecular synthesis—the deliberate combination of different discrete molecular building blocks within periodic crystalline materials. Why be concerned with the assembly of different molecular components within a solid? Broadly speaking, the interplay and communication between molecules within a solid framework provide many of the properties that are uniquely characteristic of a pure compound. The ability to change key physical properties e.g. solubility, crystal morphology, mechanical stability, etc., of a specialty chemical while retaining its essential bio-physical or molecular activity is of enormous commercial and fundamental interest. Examples of how this can be achieved are regularly demonstrated through the conversion of a pharmacologically active molecule into its chloride or acetate salt.54 Access to reliable and versatile synthetic strategies for co-crystals can furnish alternative approaches to materials design and preparation that may offer better control (and tunability) of fundamental physical properties of a wide range of specialty chemicals.
Synthesis of co-crystals and the supramolecular yield
The fact that 4-bromo-4′-cyanobiphenyl55 and 4-bromobenzonitrile56 form crystal structures where the molecular components are aligned in a head-to-tail fashion with relatively short Br⋯N contacts indicates that there are stabilizing intermolecular interactions between cyano and bromo moieties,57Fig. 4. However, there are no known examples of successful synthesis of binary co-crystals driven by CN⋯Br interactions.58 Such interactions can organize molecules within a lattice but have yet to bring about the assembly of heteromeric co-crystals.59 There is clearly a difference between observing a large number of short-contacts in molecular crystal structures composed of only one type of building block and translating such interactions into useful synthetic tools for constructing heteromeric architectures. | ||
| Fig. 4 A chain of molecules organized in a head-to-tail manner in 4-bromobenzonitrile. | ||
Much current work in organic crystal engineering is now geared towards synthesizing co-crystals using supramolecular reactions based upon reliable synthons and, so far, the majority of organic molecular co-crystals have been assembled via conventional (stronger) hydrogen bonds. Weaker hydrogen bonds and many other intermolecular interactions such as, nitro⋯iodo, cyano⋯nitro, halogen⋯halogen, etc., have not yet been found to be broadly useful tools for construction of co-crystals. The success and efficiency for any set of supramolecular reactions can be judged by the frequency of occurrence of desired intermolecular interactions and connectivities in the resulting solid. The probability that a certain motif will appear in a crystalline lattice is, in many ways, a measure of the yield of a supramolecular reaction. Just as a covalent synthetic chemist searches for ways in which a specific reaction can be promoted or prevented, a supramolecular chemist tries to identify the experimental regime where a synthon prevails despite competition from other non-covalent forces.
Heteromeric interactions are better than homomeric interactions
A survey of hydrogen-bonded co-crystals in the CSD60 reveals that most of them have been prepared using strategies that utilize suitable combinations of chemical entities (or functional groups) located on different molecules such that they would prefer to interact and bind heteromerically, Scheme 4, rather than with themselves (homomerically).61 | ||
| Scheme 4 Four dimeric motifs constructed via heteromeric intermolecular interactions. | ||
The most widely used synthons for the directed assembly of binary co-crystals have contained a carboxylic acid in combination with a suitable N-containing heterocycle. For example, there are three co-crystals in the CSD with pyrazine,62 seven with phenazine,63 sixteen with 4,4′-bipyridine,64 one with pyrimidine,65 and nine co-crystals with either azapyrine, quinoline, phenanthroline,66 and a benzoic acid–based counterpart. In every case, the expected/intended carboxylic acid⋯N(heterocycle), O–H⋯N, hydrogen bond is present. For slightly more complex heterocycles (i.e. with added substituents capable of hydrogen bonding) the results are still very consistent; 11 of 12 carboxylic acid : isonicotinamide co-crystals contain an acid⋯pyridine interaction67—a good example of a high-yielding supramolecular reaction.
There are, in fact, very few occurrences of binary hydrogen-bond based co-crystals that do not contain a primary intermolecular interaction that heteromerically link discrete building blocks. Two such examples include 4-nitrobenzamide : pyrazinecarboxamide (1 : 1) 1,68 and 3,5-dintrobenzoic acid : 4-(N,N-dimethylamino)benzoic acid (1 : 1).69 The latter was the only example, in a series of ten acid⋯acid co-crystals, where two homomeric dimers were formed in preference to one heteromeric dimer.70
The overriding conclusion from the extensive data available in the CSD is clear. In order to convince two different discrete chemical species to coexist in a molecular co-crystal there needs to be some specific molecular-recognition based reason for their solid-state union.68 Although individual structures that defy rationalization will appear from time to time, there is no doubt that the important ‘big picture’ reveals structural trends, patterns of behavior, and reproducible motifs that, when combined, can be developed into a library of high-yielding supramolecular reactions.
Do polymorphic compounds make good co-crystallizing agents?36
A good co-crystallizing agent (CA) should be able to form heteromeric intermolecular interactions with the target molecule that are more favorable than the homomeric interactions that may exist.18 The CA must also have the ability to ‘tolerate’ the presence of a different molecular building block within the same crystalline lattice, which makes it reasonable to search for co-crystallizing agents amongst polymorphic compounds.71 Polymorphism means that a compound is found in more than one crystalline manifestation which,72 in turn, indicates that such compounds display a degree of structural flexibility. In other words, the multidimensional potential-energy surface that describes the thermodynamics governing the molecular recognition processes that eventually leads to crystal growth is likely to contain many accessible local energy minima.The suggestion that polymorphic compounds are more likely to form co-crystals than compounds that never display polymorphism is somewhat difficult to quantify. However, from a practical perspective it would be extremely useful to have access to reliable guidelines for how we might focus and target a search for reliable co-crystallizing agents for a specific family of compounds. This challenge prompted us to examine four polymorphic compounds:36 isonicotinamide 2, 2-amino-5-nitropyrimidine733, 4-chlorobenzamide 4,74 and maleic hydrazide 5,75,76 and several co-crystals thereof. The reason for selecting 2–5 (apart from the fact that they are all polymorphic) was that they are all potentially capable of engaging in several well-defined and robust intermolecular hydrogen-bond interactions, Scheme 5.
 | ||
| Scheme 5 Molecules capable of forming a variety of hydrogen-bonded synthons: isonicotinamide, 2, 2-amino-3-nitropyridine, 3, 4-chlorobenzamide, 4, and maleic hydrazide, 5. | ||
Three of the four polymorphic compounds examined in this study36 readily form co-crystals, e.g. 2-hexeneoic acid : isonicotinamide, 2-amino-5-nitropyrimidine : 2-amino-3-nitropyridine, and 3-dimethylaminobenzoic acid : 4-chlorobenzamide. The structural behavior of 2–4 supports the suggestion that polymorphs make good co-crystallizing agents (provided that solubility differences are not too large). However, we were unsuccessful in preparing any co-crystals with maleic hydrazide, despite the fact that it exists in three different polymorphs. We tried a range of crystallization techniques as well as an extensive series of molecular compounds that, in principle, could form complementary hydrogen-bond interactions with a 2-pyridone moiety, Scheme 6.
 | ||
| Scheme 6 Three examples of known synthons that could facilitate the formation of co-crystals involving maleic hydrazide. | ||
Synthon flexibility
The failure to produce any co-crystals with 5 may cast doubts on the notion that polymorphic compounds make good co-crystallizing agents, unless there is a significant structural difference in the polymorphic behavior between 2–5. We decided to take a closer look at all the polymorphs 2ab, 3a–c, 4a–c, and 5a–c using graph-set notation in order to uncover possible reasons for the unwillingness of 5 to participate in molecular co-crystals. The two polymorphs of isonicotinamide, 2a and 2b, display very different intermolecular connectivities. In 2a there is an amide⋯amide head-to-head interaction as well as the commonly occurring N–H⋯O link between adjacent dimers resulting in a C(4)R22(8) motif. However, the primary intermolecular forces in 2b are significantly different as there is a catemeric amide⋯amide hydrogen bond, C(4), as well as an amide⋯pyridine interaction, C(7). A classification of the three polymorphs of 3 reveals that the expected intramolecular amine⋯nitro hydrogen bond is present in all three forms. 3b and 3c have identical intermolecular connectivities and the primary hydrogen-bond motifs are thus best described with the same graph-set notion, R22(12)R22(8)S(6). In 3a, however, there are infinite hydrogen-bonded ribbons, yielding the graph-set notation R22(10)R22(8)S(6). In the structures of 4a and 4c the amide moieties form amide–amide catemers, C(4)R22(8)R24(8). The difference between the two structures lies in the arrangement of aromatic rings with respect to one another. While the graph set notation for 4b is similar to 4a and 4c, the larger hydrogen-bonded ring creates a two-dimensional network, C(4)R22(8)R46(16). In contrast, all three polymorphs of 5, the one compound that would not readily produce co-crystals, contain identical hydrogen-bonded ladders: R24(14)R22(8), Fig. 5. | ||
| Fig. 5 The primary hydrogen-bond motifs in the three known polymorphs of maleic hydrazide display the same connectivity. | ||
The fact that these three polymorphs all contain the same extended networks was also noted in the original paper: “Three forms of MH (maleic hydrazide) provide a unique example of polymorphic structures built of similar hydrogen-bonded aggregates. The same hydrogen-bonded supramolecular aggregates organize into different lattices of different symmetries…”.76b
A detailed structural examination of 2–5 has enabled the identification of significant differences, in terms of molecular recognition patterns, between the different polymorphs that, in turn, may provide practical guidelines for ranking candidates that are potentially suitable as co-crystallizing agents. Co-crystals are not necessarily easy to prepare since such heteromeric systems are only likely to appear if the non-covalent forces between different molecules are more favorable than those that exist between molecules in the corresponding homomeric crystals. An important consideration when attempting to prepare co-crystals is to choose a co-crystallizing agent that is already known to be polymorphic. However, structural flexibility alone is not always enough. It may be equally important to select molecules that can adopt alternative packing patterns as well as display synthon flexibility—an ability to participate in several different robust and well-defined intermolecular interactions that can satisfy the demands of multiple hydrogen-bond donors/acceptors on a variety of molecules.
Beyond binary co-crystals: The need for supramolecular reagents
Through systematic structural studies of families of molecular solids we and many others have sought to examine the competition and balance between different intermolecular interactions.77–79 It is becoming clear that under certain conditions it is possible to carefully manipulate the way in which molecules recognize and bind by ‘tuning’ the strengths of site-specific complementary hydrogen-bond functionalities. We have tried to forge some of this knowledge about intermolecular forces into a practical tool for making ternary and higher-order co-crystals. Central to this approach is the availability of molecular building blocks that can provide supramolecular directionality, selectivity and reliability. Such building blocks, supramolecular reagents (SR’s), contain two or more different binding sites, attachment points, that can be used to selectively attract, bind and organize two or more different molecules into a supermolecule with predictable connectivity and shape. The supramolecular reagent is the hub for the assembly process and will become part of the supermolecule—this heteromeric aggregate will also appear in the resulting crystalline solid enabling the synthesis of co-crystals of increasing complexity.In order to establish how far this relatively simple modular supramolecular concept can be extended, we have attempted to design ternary supermolecules with predictable connectivity and stoichiometry,80Scheme 7.
 | ||
| Scheme 7 General description of a coupling reaction of two different molecules and a supramolecular reagent (SR) resulting in 1 : 1 : 1 ternary supermolecules. | ||
Since hydrogen bonds frequently form in a hierarchical fashion (best-donor to best-acceptor, second best-donor to second best-acceptor, etc.),81,82 the chances of producing a new binary co-crystal are greatly improved by positioning the best hydrogen-bond donor and the best hydrogen-bond acceptor on different molecular building blocks.68 The physical validity of a best-donor/best-acceptor classification or interpretation of hydrogen-bond interactions can be rationalized in terms of a desire of the system to maximize multiple electrostatic interactions. The centerpiece of our first deliberate attempt at making ternary co-crystals using a best-donor : best-acceptor approach was isonicotinamide.80 This molecule readily recognizes and binds to carboxylic acids to form 1 : 1 binary co-crystals that contain a robust and reproducible heteromeric hydrogen-bonded motif, Fig. 6.
 | ||
| Fig. 6 Primary motif typical of the majority of 1 : 1 isonicotinamide : carboxylic acid co-crystals.18 | ||
The best donor (carboxylic acid) and the best acceptor (pyridine nitrogen) form an intermolecular O–H⋯N hydrogen bond, and the tetrameric supermolecule is completed through a self-complementary amide–amide interaction. Isonicotinamide is a good example of a supramolecular reagent suitable for constructing ternary co-crystals: it has two distinctly different, yet relatively strong, hydrogen-bond moieties, it shows good ‘structural flexibility’ since it is polymorphic, and it also displays ‘synthon flexibility’ as the two known polymorphs display different hydrogen-bond interactions. The primary hydrogen bond in this system is the acid–pyridine interaction and, since hydrogen bonds have large electrostatic components, the strength of this interaction is governed by the acidity of the carboxylic acid donor.83
The ‘weaker’ link in the tetrameric motif shown in Fig. 6 is the amide⋯amide interaction. Since many reported structures contain heteromeric amide⋯acid hydrogen bonds (in preference to the corresponding homomeric options),84 our plan was to replace it with a more favorable heteromeric acid⋯amide interaction, allowing us to bring in a third component in a specific manner. By offering two different carboxylic acids to isonicotinamide, we expected the stronger acid to interact preferentially with the best acceptor (the pyridine nitrogen atom) and the weaker acid to form a heteromeric motif with the remaining amide moiety. This supramolecular design strategy was put to the test by allowing equimolar amounts of a weaker acid, a stronger acid, and isonicotinamide to react in an aqueous solution.
Compound 6, 3,5-dinitrobenzoic acid : isonicotinamide : 3-methylbenzoic acid (1 : 1 : 1), contains the desired three-component supermolecule with the expected connectivity. The stronger acid (pKa = 2.8)85 interacts with the pyridine nitrogen atom, and the weaker acid (pKa = 4.3) competes successfully for the amide moiety and forms a heteromeric hydrogen-bonded motif, Fig. 7.
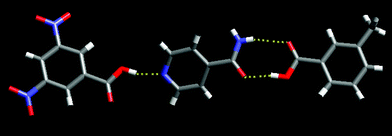 | ||
| Fig. 7 The ternary supermolecule in 6. | ||
Compound 7, 3,5-dinitrobenzoic acid : isonicotinamide : 4-(N,N-dimethyl)aminobenzoic acid (1 : 1 : 1), is also a ternary co-crystal with the intended three-component supermolecule. The acid–pyridine nitrogen interaction persists, and the weaker acid (pKa = 6.5) forms a heteromeric motif with the amide moiety, Fig. 8.
 | ||
| Fig. 8 The ternary supermolecule in 7. | ||
Finally, 3,5-dinitrobenzoic acid : isonicotinamide : 4-hydroxy-3-methoxycinnamic acid 8, is also a 1 : 1 : 1 ternary co-crystal with the same primary supramolecular connectivity as in 6 and 7, Fig. 9. The desired trimer persists even in the presence of the potentially disrupting OH-moiety on the weaker acid (pKa = 4.4).
 | ||
| Fig. 9 The ternary supermolecule in 8. | ||
All three structures, 6–8, contain ternary supermolecules synthesized with the aid of a suitable supramolecular reagent that employs a hierarchy of hydrogen-bond interactions for the desired intermolecular organization and assembly. Although these forces are weaker than covalent interactions, it is clearly possible to assemble more than two different building blocks in a preconceived manner. To date we have been able to crystallographically characterize about a dozen ternary co-crystals based upon the acid : isonicotinamide : acid combination. In each and every case, the connectivity of the observed supermolecule is consistent with the underlying supramolecular synthetic strategy and is readily rationalized through differences in pKa values—this reaction, with isonicotinamide as the supramolecular reagent seems to proceed with a very high supramolecular yield.
Despite its success, however, isonicotinamide is not sufficiently versatile to make it an ideal supramolecular reagent. First, it is capable of forming self-complementary amide⋯amide and amide⋯pyridine hydrogen bonds, which makes it inherently difficult to combine isonicotinamide with molecules that lack moieties that can compete successfully with the hydrogen-bonding capabilities of the amide functionality. Second, since the two binding sites on isonicotinamide are attached to the same backbone, it is impossible to tune the electronics of the two sites independently, which reduces the versatility. Consequently, there is a need for second-generation SR's that can be refined in such a way that they offer more opportunities for modular supramolecular synthesis of ternary co-crystals through enhanced structural selectivity and specificity.
In this context, we recently presented86 a series of asymmetric bis-heterocycles where two different binding sites (hydrogen-bond acceptor sites) are linked by a methylene bridge in order to provide increased solubility in a range of solvents. The two binding sites have significantly different basicities,87 which means that their ability to accept hydrogen bonds vary. They also lack strong hydrogen-bond donors and, consequently, homomeric intermolecular interactions will be weak and less likely to prevent the desired heteromeric interactions, Scheme 8.
 | ||
| Scheme 8 An asymmetric ditopic supramolecular reagent with two different binding sites (hydrogen-bond acceptors) that can be electronically modified independently through suitable covalent substituents. | ||
Even though pKa/pKb values do not provide direct measures of hydrogen-bond strength, hydrogen-bond abilities and free energies of complexation have been correlated with pKa values and, within closely related classes of compounds, such comparisons frequently yield correct qualitative results.88,89 The basicity of each heterocycle can also be independently altered through suitable covalent substitution, which provides a practical handle for fine-tuning differences in intermolecular reactivity. Tunability is particularly important as it creates a versatile supramolecular reagent with the potential for a high degree of transferability. The ability of these SR's to form ternary supermolecules with predictable connectivity was put to the test by allowing each SR to react with pairs of different carboxylic acids in a 1 : 1 : 1 ratio.90 The target in each case is a crystal structure containing a 1 : 1 : 1 ternary supermolecule where the primary intermolecular interactions can be rationalized according to the best donor/best acceptor protocol.
The crystal structure of 9 contains two crystallographically unique ternary supermolecules with identical connectivity, Fig. 10.
 | ||
| Fig. 10 One of the two ternary supermolecules in the crystal structure of 9. The stronger acid binds to the stronger base (left-hand side), and the second-best acid binds to the second-best base (right-hand side). | ||
The primary intermolecular interactions in this structure are the O–H⋯N hydrogen bonds between the stronger acid (3,5-dinitrobenzoic acid) and the more basic nitrogen atom of the benzimidazol-1-yl ring, and the O–H⋯N hydrogen bonds from the weaker acid (4-nitrobenzoic acid) to the less basic nitrogen atom of the pyridyl moiety.
The crystal structure of 10 shows the presence of a ternary 1 : 1 : 1 supermolecule with the same connectivity as that found in 9, Fig. 11.
 | ||
| Fig. 11 The ternary supermolecule in the crystal structure of 10. The stronger acid binds to the stronger base (left-hand side), and the weaker acid binds to the weaker base (right-hand side). | ||
The primary synthons comprise an O–H⋯N hydrogen bond between the stronger acid, 3,5-dinitrobenzoic acid, and the most basic heterocyclic moiety, and a second O–H⋯N interaction between the weaker acid, 3-N,N-dimethylaminobenzoic acid, and the less basic acceptor, the pyridyl moiety.
The crystal structure of 11 contains a 1 : 1 : 1 supermolecule with the desired connectivity, Fig. 12. The best acceptor, the benzimidazol-1-yl moiety, forms an O–H⋯N hydrogen bond with the best donor, the stronger acid. The second-best acceptor, the pyridyl moiety, binds to the weaker acid via an O–H⋯N hydrogen bond.
 | ||
| Fig. 12 The ternary supermolecule in the crystal structure of 11. The stronger acid binds to the stronger base (left-hand side), and the second-best acid binds to the second-best base (right-hand side). | ||
All three structures, 9–11, contain ternary supermolecules constructed through the deliberate use of directional intermolecular synthetic operations. The supramolecular reagents each have two binding sites that differ primarily in their basicity, but neither site is otherwise biased or predisposed towards interacting preferentially with either of the two competing carboxylic acids. The differences in basicity are translated into supramolecular reactivity and selectivity that subsequently carry over into the solid state, which demonstrates that supramolecular assembly can be controlled by fine-tuning individual binding sites. This raises the possibility that a solution to the problem of making non-covalent one-pot synthesis “sequential” may be to devise modular assembly processes based upon a hierarchy of intermolecular interactions derived from molecular properties and structural trends.
Codicil
Even though molecular recognition is typically associated with molecules in solution, such interactions are also responsible for organizing molecules in the solid state. Translating principles of molecular recognition to solid-state assembly of heteromeric molecular solids is of key importance to the development of versatile and reliable strategies for practical supramolecular synthesis. In this article we have attempted to outline some modular and transferable supramolecular design strategies based upon a hierarchy of intermolecular interactions. The starting point for these efforts is provided by a body of readily available structural information that subsequently allows for the identification of suitable supramolecular reagents. The effectiveness of these compounds as active and reliable builders of heteromeric supramolecular aggregates can eventually be evaluated and quantified using the supramolecular yield, Scheme 9. | ||
| Scheme 9 Design, implementation, and evaluation of supramolecular synthesis of co-crystals. | ||
The supramolecular synthetic strategies presented herein will, without a doubt, generate several unexpected results; however, through covalent synthesis we have unlimited opportunities for modulating the electronic and geometric details of each binding site on a supramolecular reagent such that a variety of chemical functionalities can be targeted for binding. In this way we can build a team of SR's where each member is capable of affecting the assembly of new supermolecules with a high degree of specificity and reliability, thereby clearing a path towards practical and transferable guidelines for versatile supramolecular synthesis. We are currently probing the limits and limitations of this hierarchical approach to non-covalent synthesis by examining the structural reactivity of libraries of supramolecular reagents containing multiple binding sites with easily adjustable differences in hydrogen-bond donating/accepting capabilities. Given the dramatic developments in supramolecular chemistry over the recent decades it is inevitable that much more complex supermolecules and higher-order co-crystals will be synthesized through an improved awareness of the balance and competition between intermolecular interactions. Such progress will undoubtedly also deepen our understanding of a range of fundamental events such as biomolecular activity, crystallization processes, and physico-chemical signaling mechanisms. In addition, supramolecular reactions of this type may have important ramifications for the pharmaceutical industry91 (circumventing patents, modifying crystal habit and bioavailability, and facilitating processing), and may also lead to new strategies for chiral separation.
Acknowledgements
We are grateful for financial support from NSF (CHE-0316479) and Kansas State University.References and notes
- J. D. Dunitz, in Perspectives in Supramolecular Chemistry: The Crystal as a Supramolecular Entity, ed. G.R. Desiraju, Wiley, Amsterdam, 1995 Search PubMed.
- E. J. Corey, Chem. Soc. Rev., 1988, 17, 111 RSC; K. C. Nicolaou and E. J. Sorensen, Classics in total synthesis, Wiley-VCH, Weinheim, 1996 Search PubMed.
- J.-M. Lehn, Supramolecular Chemistry, VCH, Weinheim, 1995 Search PubMed.
- C. B. Aakeröy, Acta Crystallogr., Sect. B, 1997, 53, 569 CrossRef; B. Moulton and M. J. Zaworotko, Chem. Rev., 2001, 101, 1629 CrossRef CAS; G. R. Desiraju, Acc. Chem. Res., 2002, 35, 565 CrossRef CAS; M. W. Hosseini, CrystEngComm, 2004, 6, 318 RSC; L. R. MacGillivray, CrystEngComm, 2004, 6, 77 RSC; D. Braga, Chem. Commun., 2003, 2751 RSC; L. Brammer, Chem. Soc. Rev., 2004, 33, 476 RSC; C. B. Aakeröy and A. M. Beatty, Aust. J. Chem., 2001, 54, 409 CrossRef CAS.
- D. L. Caulder and K. N. Raymond, Acc. Chem. Res., 1999, 32, 975 CrossRef CAS; D. N. Reinhoudt and M. Crego-Calama, Science, 2002, 295, 2403 CrossRef CAS; J.-M. Lehn, Science, 2002, 295, 2400 CrossRef CAS; M. W. Hosseini, Acc. Chem. Res., 2005, 38, 312; CrystEngComm Discussion 1; Innovation in Crystal Engineering (http://www.rsc.org/publishing/journals/ce/cecd1.asp).
- G. R. Desiraju, CrystEngComm, 2003, 5, 466 RSC; J. D. Dunitz, CrystEngComm, 5, 506 Search PubMed.
- A solid containing building blocks with formal charges is a salt, and there is no need to describe it as an adduct, molecular adduct, proton-transfer solid, molecular salt, or variations hereof.
- One could also make a case for including materials such as those prepared through co-condensation at reduced temperatures or elevated pressure in very elegant ways by Boese and co-workers: R. Boese, M. T. Kirchner, W. E. Billups and L. R. Norman, Angew. Chem. Int. Ed., 2003, 42, 1961 Search PubMed.
- Other intermolecular forces such as py⋯iodo will also be included.
- J. A. R. P. Sarma and G. R. Desiraju, J. Chem. Soc., Perkin Trans. 2, 1985, 1905 RSC.
- T. Sugiyama, J. Meng and T. Matsuura, J. Mol. Struct., 2002, 611, 53 CrossRef CAS.
- D. E. Lynch, G. Smith, D. Freney, K. A. Byriel and C. H. L. Kennard, Aust. J. Chem., 1994, 47, 1097 CAS.
- L. R. Nassimbeni, H. Su, E. Weber and K. Skobridis, Cryst. Growth Des., 2004, 4, 85 CrossRef CAS.
- R. D. B. Walsh, M. W. Bradner, S. Fleischman, L. A. Morales, B. Moulton, N. Rodrigues-Hornedo and M. J. Zaworotko, Chem. Commun., 2003, 186 RSC.
- D. E. Lynch, T. Latif, G. Smith, K. A. Byriel, C. H. L. Kennard and S. Parsons, Aust. J. Chem., 1998, 51, 403 CAS.
- L. R. MacGillivray, J. L. Reid and J. A. Ripmeester, J. Am. Chem. Soc., 2000, 122, 7817 CrossRef CAS.
- J. Zhu and J.-M. Zheng, Chin. J. Struct. Chem., 2004, 23, 417 Search PubMed.
- C. B. Aakeröy, A. M. Beatty and B. A. Helfrich, J. Am. Chem. Soc., 2002, 124, 14425 CrossRef.
- C. B. Aakeröy, A. M. Beatty and M. Zou, Cryst. Eng., 1998, 1, 225 CrossRef CAS.
- R. F. M. Lange, F. H. Beijer, R. P. Sijbesma, R. W. W. Hooft, H. Kooijman, A. L. Spek, J. Kroon and E. W. Meijer, Angew. Chem. Int. Ed., 1997, 36, 969 CrossRef CAS.
- J. A. Zerkowski, C. T. Seto and G. M. Whitesides, J. Am. Chem. Soc., 1992, 114, 5473 CrossRef CAS.
- M. Y. Antipin, A. I. Akhmedov, Y. T. Struchkov, E. I. Matrosov and M. I. Kabachnik, Zh. Strukt. Khim., 1983, 24, 86 CAS; D. E. Lynch, G. Smith, K. A. Byriel and C. H. L. Kennard, Acta Crystallogr., Sect. C, 1993, 49, 718 CrossRef.
- M. C. Etter and T. W. Panunto, J. Am. Chem. Soc., 1988, 110, 5896 CrossRef CAS; M. C. Etter and P. W. Baures, J. Am. Chem. Soc., 1988, 110, 639 CrossRef CAS.
- (a) L. R. MacGillivray, P. R. Diamente, J. L. Reid and J. A. Ripmeester, Chem. Commun., 2000, 359 RSC; (b) G. W. V. Cave, M. J. Hardie, B. A. Roberts and C. L. Raston, Eur. J. Org. Chem., 2001, 3227 CrossRef CAS; (c) For a good overview see: D. R. Turner, A. Pastor, M. Alajarin and J. W. Steed, Struct. Bond., 2004, 108, 97–168 Search PubMed.
- K. Kobayashi, T. Shirasaka, K. Yamaguchi, S. Sakamoto, E. Horn and N. Furukawa, Chem. Commun., 2000, 41 RSC.
- C. Glidewell, G. Ferguson, R. M. Gregson and A. J. Lough, Acta Crystallogr., Sect. C, 1999, 55, 2133 CrossRef.
- A. Ranganathan, V. R. Pedireddi and C. N. R. Rao, J. Am. Chem. Soc., 1999, 121, 1752 CrossRef CAS.
- A. De Santis, A. Forni, R. Liantonio, P. Metrangolo, T. Pilati and G. Resnati, Chem. Eur. J., 2003, 9, 3974 CrossRef CAS.
- R. B. Walsh, C. W. Padgett, P. Metrangolo, G. Resnati, T. W. Hanks and W. T. Pennington, Cryst. Growth Des., 2001, 1, 165 CrossRef CAS.
- F. Garcia-Tellado, S. J. Geib, S. Goswami and A. D. Hamilton, J. Am. Chem. Soc., 1991, 113, 9265 CrossRef.
- J.-M. Lehn, M. Mascal, A. DeCian and J. Fischer, Chem. Commun., 1990, 479 RSC.
- M. C. Etter and D. A. Adsmond, Chem. Commun., 1990, 589 RSC.
- M. Tremayne and C. Glidewell, Chem. Commun., 2000, 2425 RSC.
- O. Ermer and A. Eling, J. Chem. Soc., Perkin Trans. 2, 1994, 925 RSC.
- C. B. Aakeröy, J. Desper, B. Leonard and J. F. Urbina, Cryst. Growth Des., 2005, 5, 865 CrossRef.
- C. B. Aakeröy, A. M. Beatty, B. A. Helfrich and M. Nieuwenhuyzen, Cryst. Growth Des., 2003, 3, 159 CrossRef.
- T.-J. M. Luo and G. T. R. Palmore, Cryst. Growth Des., 2002, 2, 337 CrossRef CAS.
- V. R. Pedireddi, S. Chatterjee, A. Ranganathan and C. N. R. Rao, J. Am. Chem. Soc., 1997, 119, 10867 CrossRef CAS.
- T. L. Nguyen, F. W. Fowler and J. W. Lauher, J. Am. Chem. Soc., 2001, 123, 11057 CrossRef CAS.
- J. Xiao, M. Yang, J. W. Lauher and F. W. Fowler, Angew. Chem. Int. Ed., 2000, 39, 2132 CrossRef CAS.
- F. H. Beijer, R. P. Sijbesma, J. A. J. M. Vekemans, E. W. Meijer, H. Kooijman and A. L. Spek, J. Org. Chem., 1996, 61, 6371 CrossRef CAS.
- B.-Q. Ma and P. Coppens, Chem. Commun., 2003, 2290 RSC.
- J. J. Kane, R.-F. Liao, J. W. Lauher and F. W. Fowler, J. Am. Chem. Soc., 1995, 117, 12003 CrossRef CAS.
- P. Vishweshwar, A. Nangia and V. M. Lynch, Cryst. Growth Des., 2003, 3, 783 CrossRef CAS.
- D. S. Reddy, D. C. Craig and G. R. Desiraju, Chem. Commun., 1994, 1457 RSC.
- S. G. Fleischman, S. S. Kuduva, J. A. McMahon, B. Moulton, R. D. B. Walsh, N. Rodriguez-Hornedo and M. J. Zaworotko, Cryst. Growth Des., 2003, 3, 909 CrossRef CAS.
- T. Dahl and O. Hassel, Acta Chem. Scand., 1970, 24, 377 CrossRef CAS.
- C. B. Aakeröy, J. Desper, E. Elisabeth, B. Helfrich, B. Levin and J. Urbina, Z. Kristallogr., 2005, 220, 325 CrossRef.
- M. C. Etter, J. C. MacDonald and J. Bernstein, Acta Crystallogr., Sect. B, 1990, 46, 256 CrossRef.
- J. Bernstein, R. E. Davis, L. Shimoni and N.-L. Chang, Angew. Chem., Int. Ed., Engl., 1995, 34, 1555 CrossRef CAS.
- G. R. Desiraju, Angew. Chem., Int. Ed., Engl., 1995, 34, 2311 CrossRef CAS.
- A. Nangia and G. R. Desiraju, Acta Crystallogr., Sect. A, 1998, 54, 934 CrossRef.
- C. P. Brock and J. D. Dunitz, Acta Crystallogr., Sect. A, 1991, 47, 854 CrossRef.
- S. E. Byrn, R. R. Pfeiffer and J. G. Stowell, Solid State Chemistry of Drugs, SSCI, Inc., West Lafayette, 1999 Search PubMed.
- P. Kronenbusch, W. B. Gleson and D. Britton, Cryst. Struct. Commun., 1976, 5, 17.
- D. Britton, J. Konnert and S. Lam, Cryst. Struct. Commun., 1977, 6, 45 CAS.
- H. Quast, Y. Gorlach, E.-M. Peters, K. Peters, H. G. von Schnering, L. M. Jackman, G. Ibar and A. J. Freyer, Chem. Ber., 1986, 119, 1801 CrossRef CAS.
- The crystal structure of tetrafluoro-1,4-diiodobenzene tetramethyl-1,4-dicyanobenzene represents a rare example of a co-crystal assembled via CN⋯I interactions: D. Britton and W. B. Gleason, Acta Crystallogr., Sect. E, 2002, 58, o1375 Search PubMed.
- There are, however, examples of co-crystals formed between 1,2-dibromotetrafluoroethane and N-heterocycles such a 1,4-dimethylpiperazine: Q. Chu, Z. Wang, Q. Huang, C. Yan and S. Zhu, New. J. Chem., 2003, 27, 1522 Search PubMed.
- F. A. Allen, Acta Crystallogr., Sect. B, 2002, 58, 380 CrossRef.
- C. Huang, L. Leiserowitz and G. M. Schmidt, J. Chem. Soc., Perkin Trans. 2, 1973, 503 RSC; F. Pan, W. S. Wong, V. Gramlich, C. Bosshard and P. Gunter, Chem. Commun., 1996, 2 Search PubMed; J. A. Zerkowski, J. C. MacDonald and G. M. Whitesides, Chem. Mater., 1997, 9, 9 CrossRef CAS; R.-F. Liao, J. W. Lauher and F. W. Fowler, Tetrahedron, 1996, 52, 3153 CrossRef CAS; N. Shan, A. D. Bond and W. Jones, Cryst. Eng., 2002, 5, 9 CrossRef CAS; V. R. Pedireddi, W. Jones, A. P. Chorlton and R. Docherty, Chem. Commun., 1996, 997 RSC; P. Vishweshwar, A. Nangia and V. M. Lynch, J. Org. Chem., 2002, 67, 556 CrossRef CAS; C. B. Aakeröy, A. M. Beatty, M. Nieuwenhuyzen and M. Zou, Tetrahedron, 2000, 56, 6693 CrossRef.
- CSD codes: OCAYUM, OCASAT, and POFPAB.
- CSD codes: GURVOE, GURVUK, LUDFUL, UNECAR, ZUPKUQ, and ZUPLAX.
- CSD codes: BEQWAV, FIHYEA, FIJCIK, FIJCUW, HUPPOX, MUFNIK, NOPXIZ, OJENIA, PULWUO, RAPHAR, SUXVOW, UDUZIC, UDOZOI, UHELUO, XUNGIW, and XUNGOC.
- CSD code: POFPEF.
- CSD codes: CUFSET, LUSWEB, PANYIM, PANZEJ, PAPDIT, PAPFOB, UNEBOE, WUKREZ, and WUKROJ.
- CSD codes: AJAKEB, AJAKIF, ASAXOH, ASAXUN, BUDWEC, BUDZUV, BUFBIP, BUFQAU, LUNMEM, LUNMIC, LUNMOW, and VAKTOR.
- C. B. Aakeröy, J. Desper and B. A. Helfrich, CrystEngComm, 2004, 6, 19 RSC.
- C. V. K. Sharma, K. Panneerselvam, T. Pilati and G. R. Desiraju, J. Chem. Soc., Perkin Trans. 2, 1993, 2209 RSC.
- V. R. Pedireddi and J. PrakashReddy, Tetrahedron Lett., 2002, 43, 4927 CrossRef CAS.
- There are, of course, other aspects that need to be considered e.g., solubility differences between drug and CA (if the co-crystals are to be prepared via solution-based methods) before embarking upon a series of co-crystallizations.
- J. Bernstein, Polymorphism in Molecular Crystals, Oxford University Press, New York, 2002 Search PubMed.
- C. B. Aakeröy, M. Nieuwenhuyzen and S. L. Price, J. Am. Chem. Soc., 1988, 120, 8986.
- Forms I and III of 4-chlorobenzamide: T. Taniguch, K. Nakata, Y. Takaki and K. Sakurai, Acta Crystallogr., Sect. B, 1978, 34, 2574 Search PubMed . Form II: T. Hayashi, K. Nakata, Y. Takaki and K. Sakurai, Bull. Chem. Soc. Jpn., 1980, 53, 801 CrossRef.
- P. D. Cradwick, Nature, 1975, 258, 774 CAS.
- (a) A. Katrusiak, Acta Crystallogr., Sect. C, 1993, 49, 36 CrossRef; (b) A. Katrusiak, Acta Crystallogr., Sect. B, 2001, 57, 697 CrossRef CAS.
- C. B. Aakeröy, A. M. Beatty and D. S. Leinen, CrystEngComm, 2002, 4, 310 RSC; J. Valdés-Martínez, M. Del Rio-Ramirez, S. Hernández-Ortega, C. B. Aakeröy and B. A. Helfrich, Cryst. Growth Des., 2001, 2, 485 CrossRef; C. B. Aakeröy, A. M. Beatty, M. Tremayne, D. M. Rowe and C. C. Seaton, Cryst. Growth Des., 2001, 2, 377 CrossRef.
- C. B. Aakeröy, A. M. Beatty and D. S. Leinen, Cryst. Growth Des., 2000, 1, 47 Search PubMed; C. B. Aakeröy, A. M. Beatty, M. Nieuwenhuyzen and M. Zou, J. Mater. Chem., 1998, 8, 1385 RSC; C. B. Aakeröy, J. Desper and J. F. Urbina, Cryst. Growth Des., 2005, 5, 1283 CrossRef.
- A. D. Burrows, Struct. Bond., 2004, 111, 55–96.
- C. B. Aakeröy, A. M. Beatty and B. A. Helfrich, Angew. Chem. Int. Ed., 2001, 40, 3240 CrossRef CAS.
- M. C. Etter, J. Phys. Chem., 1991, 95, 4601 CrossRef CAS.
- M. C. Etter, Acc. Chem. Res., 1990, 23, 120 CrossRef CAS.
- If the acid is too strong, the proton will be transferred to the nitrogen atom resulting in an ionic compound and not a co-crystal. Some examples of crystal engineering of ionic systems include e.g.: J. A. Swift, A. M. Pivovar, A. M. Reynolds and M. D. Ward, J. Am. Chem. Soc., 1998, 120, 5887 Search PubMed; F. Xue and T. C. W. Mak, J. Phys. Org. Chem., 2000, 13, 405 CrossRef CAS; P. Cudic, J.-P. Vigneron, J.-M. Lehn, M. Cesario and T. Prangé, Eur. J. Org. Chem., 1999, 2479 CrossRef CAS; C. V. K. Sharma and A. Clearfield, J. Am. Chem. Soc., 2000, 122, 4394 CrossRef CAS; S. Ferlay, R. Holakovsky, M. W. Hosseini, J.-M. Planeix and N. Kyritsakas, Chem. Commun., 2003, 1224 CrossRef CAS; C. B. Aakeröy, P. B. Hitchcock and K. R. Seddon, J. Chem. Soc., Chem. Commun., 1992, 553 RSC; C. B. Aakeröy, D. P. Hughes and M. Nieuwenhuyzen, J. Am. Chem. Soc., 1996, 118, 10134 RSC; A. M. Beatty, K. E. Granger and A. E. Simpson, Chem. Eur. J., 2002, 8, 3254 CrossRef; A. M. Beatty, C. L. Schneider, A. E. Simpson and J. L. Zaher, CrystEngComm, 2002, 4, 282 CrossRef CAS; D. Braga, L. Maini, M. Polito and F. Grepioni, Struct. Bond., 2004, 111, 1–32 RSC.
- L. Leiserowitz and F. Nader, Acta Crystallogr., Sect. B, 1977, 33, 2719 CrossRef.
- All pKa/pKb values from: G. Kartum, W. Vogel and K. Andrussov, Dissociation constants of organic acids in aqueous solution, Butterworth & Co. Publishers, London, 1961 Search PubMed.
- C. B. Aakeröy, J. Desper and J. F. Urbina, Chem. Commun., 2005, 2820 RSC.
- An estimate for the differences in basicity between the pyridyl and the benzimidazol-1-yl moieties were obtained by calculating pKa values for the conjugated acids of the two hydrogen-bond acceptors in each SR: 3-(benzimidazol-1-yl)methylpyridine: pKa = 4.71 ± 0.10 and 5.72 ± 0.30 for pyridyl and benzimidazol-1-yl moieties, respectively; 3-(2-methylbenzimidazol-1-yl)methylpyridine: pKa = 4.72 ± 0.10 and 5.85 ± 0.18 for pyridyl and benzimidazol-1-yl moieties, respectively. The calculations were performed using Advanced Chemistry Development (ACD/Labs) Software Solaris V4.76 (© 1994–2005 ACD/Labs).
- M. H. Abraham, Chem. Soc. Rev., 1993, 22, 73 RSC.
- S. Shan, S. Loh and D. Herschlag, Science, 1996, 272, 97 CrossRef CAS.
- 3,5-Dinitrobenzoic acid (pKa = 2.8); 4-nitrobenzoic acid (pKa = 3.44); 3-N,N-(dimethylamino)benzoic acid (pKa = 4.30): G. Kartum, W. Vogel and K. Andrussov, Dissociation constants of organic acids in aqueous solution, Butterworth & Co. Publishers, London, 1961 Search PubMed.
- Ö. Almarsson and M. J. Zaworotko, Chem. Commun, 2004, 1889 RSC.
| This journal is © The Royal Society of Chemistry 2005 |