Maleates from diazoacetates and dilactones from head-to-head dimerisation of alkenyl diazoacetates using Grubbs' 2nd-generation ruthenium carbene catalyst
David M.
Hodgson
* and
Deepshikha
Angrish
Department of Chemistry, University of Oxford, Chemistry Research Laboratory, Mansfield Road, Oxford, UK OX1 3TA. E-mail: david.hodgson@chem.ox.ac.uk
First published on 31st August 2005
Abstract
Grubbs' 2nd-generation ruthenium carbene catalyst homocouples diazoacetates to maleates and also catalyses head-to-head dimerisation of alkenyl diazoacetates giving dienyl dilactones.
Alkene metathesis has become a popular synthetic method.1 It relies on the ability of well-defined (pre-)catalysts [typically, commercially available Grubbs' 1st- and 2nd-generation ruthenium carbenes, (PCy3)2Cl2Ru
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CHPh (1) and 2, respectively] to effect the chemistry stereoselectively under mild conditions, with high functional group compatibility and predictable selectivity between various types of alkenes. For example, acrylates 3 dimerise to fumarates 4,2 and unsaturated acrylates 5 can be converted to macrocycles 6 by head-to-tail dimerising (or trimerising) metathesis3
(Scheme 1). In the present paper, we communicate the ability of such catalysts to generate maleates from diazoacetates, and macrocyclic dilactones by head-to-head dimerisation of unsaturated diazoacetates.
CHPh (1) and 2, respectively] to effect the chemistry stereoselectively under mild conditions, with high functional group compatibility and predictable selectivity between various types of alkenes. For example, acrylates 3 dimerise to fumarates 4,2 and unsaturated acrylates 5 can be converted to macrocycles 6 by head-to-tail dimerising (or trimerising) metathesis3
(Scheme 1). In the present paper, we communicate the ability of such catalysts to generate maleates from diazoacetates, and macrocyclic dilactones by head-to-head dimerisation of unsaturated diazoacetates.
 | ||
| Scheme 1 2,3 | ||
Transition metal-catalysed cyclopropanation of alkenes using diazo compounds has been extensively examined, with the reaction between styrene and ethyl diazoacetate (EDA) a particular focus regarding control of diastereo- and enantioselectivity.4 The homocoupling (dimerisation) of EDA to a mixture of diethyl maleate (DEM) and fumarate (DEF), usually strongly favouring the former, is often observed as an unwanted side reaction during cyclopropanation and may be minimised either by slow addition of the diazo compound to the alkene and catalyst and/or by using an excess of alkene.5 Although comparatively less well studied as a specifically desired process, several transition metal catalysts are known to homocouple α-diazocarbonyl compounds, and the transformation has found sporadic use in synthesis.6 Currently, one of the most efficient and stereoselective processes uses the half-sandwich ruthenium(II) complex (PPh3)2ClRu(η5-C5H5) (0.1 mol%, toluene or CHCl3, ∼ 60 °C) to dimerise EDA to DEM (quant., DEM ∶ DEF, > 99 ∶ 1, by 1H NMR).7 The latter catalyst also effects cyclopropanation of styrenes with EDA.8
We found that Grubbs' 2nd-generation catalyst 2 (0.5 mol%) transformed EDA (0.2 mol dm−3 in CH2Cl2, room temperature, 15 h) into mainly DEM (95% isolated yield; DEM ∶ DEF, 98 ∶ 2 by GC of crude product; Scheme 2, R = Et). No homocoupling of EDA occurred with PCy3 (0.5 mol%) by itself, and subsequent addition of catalyst 2 (0.5 mol%) only led to sluggish coupling (one-third of EDA still remained after 40 h). This latter result implicates phosphine dissociation as an important step in the homocoupling7 (as it is in metathesis1,9). Use of Grubbs' 1st-generation catalyst 1 provided a lower isolated yield of DEM (73%) and was not studied further. With a reduced loading of catalyst 2 (0.1 mol%), stereoselectivity for DEM was essentially complete10 (DEM ∶ DEF, > 99 ∶ 1), although the reaction did not proceed to completion even after 72 h, resulting in a 54% isolated yield of DEM. Isopropyl and t-butyl diazoacetates with 2 (0.5 mol%) also gave the corresponding maleates (95% and 99% isolated yields, respectively), with high stereoselectivity for maleate over fumarate (97 ∶ 3 and 99 ∶ 1, respectively).
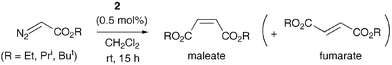 | ||
| Scheme 2 | ||
A mixture of EDA and styrene (1 ∶ 1, CH2Cl2, room temperature) was then treated with Grubbs' 2nd-generation catalyst 2 (1 mol%), to see if the latter could induce cyclopropanation. After 18 h, DEM was mainly observed along with partial (∼ 30%) conversion of styrene to trans-stilbene. That a (source of) metathetically active catalyst was still present at this point was established in a second run of the reaction in which, prior to chromatography, the reaction mixture was concentrated at ∼ 50 °C which led to all the remaining styrene being converted to stilbene (isolated in quantitative yield; 95% yield of DEM was also obtained). Sampling over 18 h during a duplicate run at room temperature indicated EDA reacted relatively rapidly (80% consumed after 2 h), whereas stilbene formed gradually (3% after 2 h, 15% after 4 h). In the absence of EDA, catalyst 2 (0.5 mol%, CH2Cl2, room temperature) converts styrene quantitatively into stilbene after 18 h. However, the presence of 1 equiv. (relative to styrene) of DEM, DEF, or MeOAc slowed stilbene formation (after 24 h, ∼ 30% with DEM or DEF, 50% with MeOAc), suggesting competition between styrene, and DEM, DEF, or MeOAc for PCy3-dissociated 2. Attempts to observe cyclopropanation with EDA and catalyst 2, by increasing the amount of styrene (to 2 equiv.) or by slow (syringe pump) addition of EDA proved fruitless. Ethyl cinnamate was also not detected in any of these experiments. It therefore appears that catalyst 2 is able to promote two different carbene transformations in the same flask, with no carbene crossover.11
A tentative catalytic cycle which rationalises the above observations concerning EDA homocoupling is outlined in Scheme 3. The coordinatively unsaturated intermediate 7 reacts with EDA to generate ester carbene 8. Ester carbene 8 does not display propensity for cyclopropanation (or metathesis), but preferentially undergoes reaction with further EDA (8 → 9 → 10). Highly diastereoselective attack by EDA on a (Cu-based) ester carbene followed by anti-elimination of the metal and N2 has been suggested as the origin of the stereoselectivity for maleate over fumarate in EDA homocoupling.12 DEM dissociation from 10 then completes the catalytic cycle. With EDA, styrene and catalyst 2, the cycle shown in Scheme 3 preferentially (but not exclusively) operates alongside the well-established metathesis catalytic cycle1,9 involving common intermediate 7 and styrene [with the difference that after one metathesis turnover a ruthenium methylidene rather than benzylidene (i.e., H rather than Ph) is involved].
 | ||
| Scheme 3 | ||
The ability of Grubbs' catalyst 2 to catalyse both diazoacetate dimerisation and alkene metathesis led us to examine head-to-head dimerisation of unsaturated diazoacetates 11 (readily available from the corresponding unsaturated alcohol and glyoxylic acid chloride Ts hydrazone),13 as a route to dienyl dilactones 12 (Scheme 4).
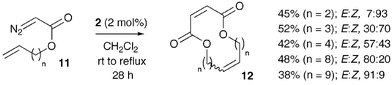 | ||
| Scheme 4 | ||
With catalyst 2 (1 mol%) and unsaturated diazoacetates 11 (0.07 mol dm−3 in CH2Cl2) it was found that maleate formation was complete after 14–18 h at room temperature, but only a small amount of alkene metathesis had occurred. This presumably reflects the lower metathesis activity of alkyl-substituted terminal alkenes compared with styrene. Thus, an additional 1 mol% of catalyst 2 was added and the reaction heated to reflux to promote ring-closing metathesis.†cis-Alkenes14 were preferentially formed by metathesis when leading to 12- and 14-membered dilactones, whereas trans stereochemistry was increasingly favoured for larger rings (16–26-membered).
In summary, Grubbs' 2nd-generation ruthenium carbene is shown to act as an efficient catalyst for highly stereoselective homocoupling of diazoacetates; in the presence of additional alkene functionality cyclopropanation is not observed but rather metathetical activity is retained, and can be exploited with unsaturated diazoacetates giving dienyl dilactones. The contrasting stereo- and regiochemical outcomes between (unsaturated) acrylates and diazoacetates with catalyst 2 are noteworthy. Studies with other unsaturated diazocompounds are under investigation.
We thank the University of Oxford for a full Clarendon Fund bursary award (to D. A.) and the EPSRC National Mass Spectrometry Service Centre for mass spectra.
Notes and references
- Handbook of Metathesis, ed. R. H. Grubbs, Wiley-VCH, Weinheim, 2003 Search PubMed.
- T.-L. Choi, C. W. Lee, A. K. Chatterjee and R. H. Grubbs, J. Am. Chem. Soc., 2001, 123, 10417–10418 CrossRef CAS.
- C. W. Lee and R. H. Grubbs, J. Org. Chem., 2001, 66, 7155–7158 CrossRef CAS . See also: A. Fürstner, O. R. Thiel, L. Ackermann, H.-J. Schanz and S. P. Nolan, J. Org. Chem., 2000, 65, 2204–2207 Search PubMed.
- H. M. L. Davies and E. G. Antoulinakis, Org. React., 2001, 57, 1–326 CAS.
- Intriguingly, homocoupling is completely suppressed using a Cu N-heterocyclic carbene system: M. R. Fructos, T. R. Belderrain, M. C. Nicasio, S. P. Nolan, H. Kaur, M. M. Díaz-Requejo and P. J. Pérez, J. Am. Chem. Soc., 2004, 126, 10846–10847 Search PubMed.
- M. P. Doyle, M. A. McKervey and T. Ye, Modern Catalytic Methods for Organic Synthesis with Diazo Compounds, John Wiley and Sons, New York, 1998, pp. 624–627 Search PubMed. For a recent example in synthesis, see: G.-Y. Li and C.-M. Che, Org. Lett., 2004, 6, 1621–1623 Search PubMed.
- (a) W. Baratta, A. Del Zotto and P. Rigo, Chem. Commun., 1997, 2163–2164 RSC; (b) W. Baratta, A. Del Zotto and P. Rigo, Organometallics, 1999, 18, 5091–5096 CrossRef CAS; (c) A. Del Zotto, W. Baratta, G. Verado and P. Rigo, Eur. J. Org. Chem., 2000, 2795–2801 CrossRef CAS.
- W. Baratta, W. A. Herrmann, R. M. Kratzer and P. Rigo, Organometallics, 2000, 19, 3664–3669 CrossRef CAS . See also: B. Çentinkaya, I. Özdemir and P. H. Dixneuf, J. Organomet. Chem., 1997, 534, 153–158 Search PubMed.
- M. S. Sanford, J. A. Love and R. H. Grubbs, J. Am. Chem. Soc., 2001, 123, 6543–6554 CrossRef CAS.
- E. Graban and F. R. Lemke, Organometallics, 2002, 21, 3823–3826 CrossRef CAS.
- For competitive cyclopropanation and cross-metathesis with EDA and alkenes using Ru2(OAc)4, see: A. F. Noels, A. Demonceau, E. Carlier, A. J. Hubert, R. L. Márquez-Silva and R. A. Sánchez-Delgado, J. Chem. Soc., Chem. Commun., 1988, 783–784 Search PubMed.
- D. S. Wulfmann, B. W. Peace and R. S. McDaniel, Jr., Tetrahedron, 1976, 32, 1251–1255 CrossRef CAS . See also: T. Oshima and T. Nagai, Tetrahedron Lett., 1980, 21, 1251–1254 Search PubMed and refs. 7b and 10 for additional suggestions.
- E. J. Corey and A. G. Myers, Tetrahedron Lett., 1984, 25, 3559–3562 CrossRef CAS; M. P. Doyle and I. M. Phillips, Tetrahedron Lett., 2001, 42, 3155–3158 CrossRef CAS.
- A. Fürstner and K. Langemann, Synthesis, 1997, 792–803 CrossRef CAS.
Footnote |
| † Typical procedure for dilactone formation: Grubbs' 2nd-generation catalyst 2
(3 mg, 3.5 µmol) was added to a stirred solution of but-3-enyl diazoacetate 11
(n
= 2)
(49 mg, 0.35 mmol) in CH2Cl2
(5 cm3) at rt. After 14 h, further catalyst 2
(3 mg, 3.5 µmol) was added and the reaction mixture heated to reflux for 14 h. The mixture was then concentrated under reduced pressure and purified by flash chromatography (SiO2, 5 → 10% EtOAc in hexane) to afford a yellow oil, dilactone 12
(n
= 2)
(15.5 mg, 45%, E
∶
Z 7 ∶ 93); Rf 0.56 (50% EtOAc in hexane); νmax(neat)/cm−1 2962w, 1794s, 1382w, 1261m and 1100s; δH(400 MHz; CDCl3)
Z-isomer: 6.18 (1H, s, CHCO), 5.62–5.54 (1H, m, CHCH2), 4.32–4.24 (2H, m, OCH2) and 2.51 (2H, q, J 5.5, CHCH2); discernible data for E-isomer: 6.16 (1H, s, CHCO), 5.42–5.38 (1H, m, CHCH2) and 2.46–2.38 (2H, m, CHCH2); δC(100 MHz, CDCl3)
Z-isomer: 165.1 (CO), 128.8 (CH), 128.7 (CH), 64.8 (OCH2) and 26.4 (CHCH2); discernible data for E-isomer: 129.2 (CH), 63.3 (OCH2) and 32.5 (CHCH2); m/z
(ES) 197 (M + H+, 30%), 214 (100) and 215 (10); Found M + H, 197.0816. C10H13O4 requires M 197.0814. |
| This journal is © The Royal Society of Chemistry 2005 |