Organocatalytic asymmetric α-bromination of aldehydes and ketones†
Søren
Bertelsen
,
Nis
Halland
,
Stephan
Bachmann
,
Mauro
Marigo
,
Alan
Braunton
and
Karl Anker
Jørgensen
*
Danish National Research Foundation: Center for Catalysis, Department of Chemistry, Aarhus University, Aarhus C, DK-8000, Denmark. E-mail: kaj@chem.au.dk; Fax: 45 8619 6199; Tel: 45 8942 3910
First published on 30th August 2005
Abstract
The first organocatalytic enantioselective α-bromination of aldehydes and ketones is presented; a C2-symmetric diphenylpyrrolidine catalyst afforded the α-brominated aldehydes in good yields and up to 96% ee, while ketones were α-brominated by a C2-symmetric imidazolidine in up to 94% ee; furthermore, the organocatalytic enantioselective α-iodination of aldehydes is also demonstrated to proceed with up to 89% ee.
The transformation of C–H into C–X (X = F, Cl, Br, I) bonds with stereochemical control of the chiral carbon center formed is an important challenge in organic and medicinal chemistry.1
In recent years a number of enantioselective halogenation reactions have been developed using chiral Lewis acids and organic compounds2 as the catalysts. For the chiral Lewis acid-catalyzed reactions the substrates are mainly β-keto esters and phosphonates using electrophilic halogenation reagents and enantioselective α-fluorination, α-chlorination and α-brominations have been successfully developed.3 A chiral phase-transfer catalyst derived from a cinchona alkaloid has also been shown to be effective for the α-fluorination of β-keto esters giving the corresponding optically active α-fluorinated compounds with up to 69% ee.4
Optically active α-chloro- and α-bromoesters have been obtained from ketenes which are formed in situ from acetyl chlorides and base, followed by treatment with an electrophilic chlorine or bromine source, in the presence of a cinchona alkaloid acting as a chiral nucleophilic catalyst.5
Recently, the organocatalytic enantioselective α-chlorination and α-fluorination of aldehydes, and α-chlorination of ketones were described. Two papers independently presented the α-chlorination of aldehydes. MacMillan et al. used a chiral imidazolidinone as the catalyst and hexachloro-cyclohexadienone as the chlorine source leading to the α-chlorinated aldehydes in high yield and enantioselectivity (92–95% ee).6 Our approach was based on NCS as the chlorinating reagent and L-proline amide or C2-symmetric diphenylpyrrolidine as the catalysts. The latter afforded the highest enantiomeric excess of the α-chlorinated aldehydes (94–97% ee) in high yields.7 For the direct α-chlorination of ketones, a simple extension of the related aldehyde transformation was not possible. Neither proline, nor the optimal catalysts for the chlorination of aldehydes promoted this reaction efficiently.8 A thorough screening of a number of organocatalysts led to the use of a C2-symmetric imidazolidine as the catalyst of choice.
Four papers were very recently published within a few weeks presenting the organocatalytic enantioselective α-fluorination of carbonyl compounds.9 In the paper by Enders et al.,9aL-proline and derivatives were shown to catalyze the α-fluorination of e.g. hexanal and cyclohexanone in moderate to good yields and up to 36% ee using Selectfluor as the fluorinating agent. We have developed a highly enantioselective α-fluorination of aldehydes employing NFSI as the fluorine source and only 1 mol% of a silyl-protected proline-derived catalyst.9b This system led to the formation of stereogenic C–F centers with up to 97% ee. Barbas et al.9c and MacMillan et al.9d have employed an imidazolidinone catalyst and NFSI, and obtained α-fluorinated aldehydes with high optical purity. However, high catalyst loadings (20–100 mol%) were employed.
In this communication the first organocatalytic enantioselective α-bromination of aldehydes and ketones is presented (Scheme 1).

We initially screened several bromine sources 2a–c for the α-bromination of 3-methyl butanal 1a in the presence of various chiral amines as catalysts (Scheme 2).
 | ||
| Scheme 2 Bromination reagents and catalysts screened during the optimization. | ||
We started our investigations using the reaction conditions successfully applied to the α-chlorination of aldehydes with NBS (2a) as the bromine source.7 However, these conditions were found to be unsuitable, giving low conversion and enantioselectivity (8% yield and 19% ee). Further studies indicated that this might be due to the increased reactivity of NBS 2a compared to that of NCS, and hence the temperature was lowered to −24 °C which gave 91% conversion of 3-methyl butanal 1a affording 2-bromo-3-methyl butanal 4a in 49% ee (Table 1, entry 1). A further increase in enantioselectivity was observed when 2-benzhydryl-pyrrolidine 3b was applied as the catalyst and when 20 mol% benzoic acid was added to the reaction mixture full conversion was observed within 1 h (entry 2). Interestingly, both the yield and enantioselectivity were significantly lower in the absence of an acid additive (entry 3). Compound 3c, a highly efficient catalyst for the enantioselective α-chlorination of aldehydes, gave good conversion when NBS 2a was employed as the bromine source (entry 4). However, application of the easily synthesized, air-stable 4,4-dibromo-2,6-di-tert-butyl-cyclohexa-2,5-dienone102b improved the enantioselectivity and 2b was found to be an excellent reagent compared to the other bromine sources (entries 4, 5, 7). The reaction conditions were optimized using (2R,5R)-diphenylpyrrolidine 3c as catalyst and 2b as bromine source and the yield was found to be strongly solvent dependent (entries 8–11). We were pleased to find high enantioselectivity and conversion in a 1 ∶ 1 mixture of CH2Cl2 and pentane (entry 11). Furthermore, this mixture prevented racemization of the optically active product, since the enantiomeric excess of 4a was unaltered after 2 days. It is notable that the catalysts 3b and 3c gave the opposite enantiomer of 4a, compared to L-proline amide 3a.
| Entry | Cat | Halogen source | Solvent | Additive (mol%) | Conversiona (%) | eeb (%) |
|---|---|---|---|---|---|---|
| a Conversion after 60 min measured by 1H NMR spectroscopy of the crude reaction mixture and confirmed by GC. b ee of 2-bromo-3-methyl butanal determined by CSP-GC. c 10 mol% catalyst used compared to 20 mol% in the other experiments. | ||||||
| 1 | 3a | 2a | CH2Cl2 ∶ pentane 1 ∶ 3 | 91 | −49 | |
| 2 | 3b | 2a | CH2Cl2 ∶ pentane 1 ∶ 3 | PhCO2H (20) | 100 | 58 |
| 3 | 3b | 2a | CH2Cl2 ∶ pentane 1 ∶ 3 | 24 | 11 | |
| 4 | 3c | 2a | CH2Cl2 ∶ pentane 1 ∶ 3 | PhCO2H (20) | 95 | 45 |
| 5 | 3c | 2b | CH2Cl2 ∶ pentane 1 ∶ 3 | PhCO2H (20) | 71 | 97 |
| 6c | 3c | 2b | CH2Cl2 ∶ pentane 1 ∶ 3 | PhCO2H (10) | 60 | 97 |
| 7 | 3c | 2c | CH2Cl2 ∶ pentane 1 ∶ 3 | PhCO2H (20) | 81 | 86 |
| 8 | 3c | 2b | PhMe | PhCO2H (20), H2O (200) | 40 | 94 |
| 9 | 3c | 2b | Pentane | PhCO2H (20), H2O (200) | 46 | 93 |
| 10 | 3c | 2b | MeCN | PhCO2H (20), H2O (200) | 100 | 83 |
| 11 | 3c | 2b | CH2Cl2 ∶ pentane 1 ∶ 1 | PhCO2H (20), H2O (200) | 90 | 96 |
After optimizing the reaction conditions we expanded the scope of the reaction by α-brominating different aldehydes (Scheme 3 and Table 2).
 | ||
| Scheme 3 Organocatalytic α-bromination of aldehydes. | ||
 | ||
| Scheme 4 Organocatalytic α-bromination of different ketones. | ||
| Entry | R | Isolated yield (%)a | eeb (%) |
|---|---|---|---|
| a Isolated yield of the corresponding alcohol after NaBH4 reduction. b ee determined of the α-bromo aldehydes by CSP-GC. c −24 °C, 1.3 equiv. of 2c. | |||
| 1c | i-Pr–1a | 87–5a | 96 |
| 2c | t-Bu–1b | 94–5b | 89 |
| 3 | Et–1c | 72–5c | 77 |
| 4 | n-Pr–1d | 82–5d | 85 |
| 5 | n-Hex–1e | 95–5e | 68 |
| 6 | Cyclohexyl–1f | 92–5f | 73(S) |
| 7 | Allyl–1g | 74–5g | 76 |
The enantioselective α-bromination proceeded well for aldehydes 1a–g with isolated yields of the α-bromo alcohols 5a–g in the range of 72–95% in 2 steps (Table 2). Furthermore, good to excellent enantioselectivity, in the range 68–96% ee, were observed for linear, branched, cyclic and unsaturated aldehydes (entries 1–7). The absolute configuration of the chiral carbon center formed has been assigned to be (S) by comparison of the optical rotation of bromoalcohol 5f with literature values,11 when using the (2R,5R)-diphenylpyrrolidine 3c as catalyst. This is the same absolute configuration found in the α-chlorination of aldehydes using NCS and the same catalyst.7
The reaction condition for the organocatalytic α-bromination of aldehydes have been applied to the enantioselective α-iodination of aldehydes as well. According to our knowledge, there is no procedure for the direct α-iodination of aldehydes. Iodination of aldehydes such as 3-methyl butanal 1a with NIS 2d and 3e as the catalyst, was observed to be a very rapid reaction, with full conversion in only 20 min. For the α-iodination of 1a, 78% yield and 89% ee of 2-iodo-3-methyl butanal was obtained, while butanal 1c afforded the corresponding optically active α-iodo aldehyde in 60% ee, however, only 30% yield was obtained.
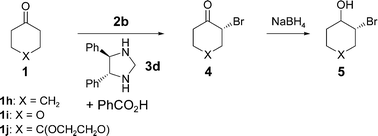
We have extended the catalytic α-bromination reaction to the α-bromination of ketones (Scheme 3). We were pleased to find that the bromine source 4,4-dibromo-2,6-di-tert-butyl-cyclohexa-2,5-dienone 2b was significantly better compared to the other reagents tested. Furthermore, it was established that the reaction conditions previously developed for the organocatalytic asymmetric chlorination of ketones8 also proved successful for the α-bromination of ketones. Table 3 entries 1–7 show the optimization of the reaction conditions for the α-bromination of cyclohexanone 1h.
| Entry | Ketone | Solvent | Temp/°C | Time/h | Conversionb (isolated yield (%)) | eec (%) |
|---|---|---|---|---|---|---|
| a Reaction conditions: See Supporting Information1. b Measured by 1H NMR of the crude reaction mixture and confirmed by GC. c ee determined of the α-bromo ketones by CSP-GC. d Yield of the corresponding cis-alcohol after NaBH4 reduction and FC. e Yield after FC on Iatrobeads. | ||||||
| 1 | 1h | MeCN | −24 | 20 | 66 (58)d–5h | 85 |
| 2 | 1h | CH2Cl2 ∶ pentane 1 ∶ 3 | −24 | 20 | 87–5h | 86 |
| 3 | 1h | Et2O | −30 | 0.5 | 16–5h | 88 |
| 4 | 1h | i-PrOH | −30 | 20 | 63–5h | 88 |
| 5 | 1h | EtOH | −30 | 20 | 66–5h | 94 |
| 6 | 1h | t-BuOMe | −30 | 40 | 90 (81)d–5h | 90 |
| 7 | 1h | THF | −30 | 20 | 76 (70)d–5h | 91 |
| 8 | 1i | THF | −30 | 20 | 80e–4i | 89 |
| 9 | 1j | THF | −30 | 40 | 67e–4j | 73 |
Cyclohexanone 1h, in the presence of catalyst 3d, could be brominated in good yield with an enantioselectivity of up to 94% ee under the optimized reaction conditions (Table 3, entry 5). For the two other cyclic ketones (1i,j) presented in Table 3, the α-bromination also procceds well and with good enantioselectivity (entries 8, 9). The absolute configuration of the chiral carbon center formed has been assigned by X-ray analysis of compound 4i to be (R) when using the (4R,5R)-diphenylimidazolidine catalyst. ‡This is the same absolute configuration as observed in the corresponding α-chlorination of ketones using the same catalyst.8
In conclusion, we have developed the first organocatalytic enantioselective α-bromination of aldehydes and ketones. For the aldehydes a C2-symmetric diphenylpyrrolidine catalyst gave the optically active α-brominated aldehydes in moderate to good yields and up to 96% ee, while the ketones were α-brominated by a C2-symmetric imidazolidine in up to 94% ee. Furthermore, we have also demonstrated the organocatalytic enantioselective α-iodination of aldehydes to proceed with up to 89% ee.
This work was made possible by a grant from The Danish National Research Foundation. We are grateful to Dr. Rita G. Hazell for performing X-ray crystallographic studies.
Notes and references
- For some recent reviews see e.g.: (a) H. Ibrahim and A. Togni, Chem. Commun., 2004, 1147 RSC; (b) K. Muniz, Angew. Chem., Int. Ed., 2001, 40, 1653 CrossRef CAS; (c) M. Oestreich, Angew. Chem., Int. Ed., 2005, 44, 2324 CrossRef CAS; (d) S. France, A. Weatherwax and T. Lectka, Eur. J. Org. Chem., 2005, 475 CrossRef CAS.
- For general reviews on organocatalysis see e.g.: (a) P. I. Dalko and L. Moisan, Angew. Chem., Int. Ed., 2004, 43, 5138 CrossRef CAS; (b) J. Seayad and B. List, Org. Biomol. Chem., 2005, 3, 719 RSC; (c) A. Berkessel and H. Gröger, Metal-Free Organic Catalysts in Asymmetric Synthesis, Wiley-VCH, Weinheim, 2004 Search PubMed.
- See e.g.: (a) L. Hintermann and A. Togni, Angew. Chem., Int. Ed., 2000, 39, 4359 CrossRef CAS; (b) L. Hintermann and A. Togni, Helv. Chim. Acta., 2000, 83, 2425 CrossRef CAS; (c) H. Ibrahim, F. Kleinbeck and A. Togni, Helv. Chim. Acta., 2004, 87, 605 CrossRef CAS; (d) Y. Hamashima, K. Yaga, H. Takano, L. Tomás and M. Sodeoka, J. Am. Chem. Soc., 2002, 124, 14530 CrossRef CAS; (e) J.-A. Ma and D. Cahard, Tetrahedron: Asymmetry, 2004, 15, 1007 CrossRef CAS; (f) M. Marigo, N. Kumaragurubaran and K. A. Jørgensen, Chem. Eur. J., 2004, 10, 2133 CrossRef CAS; (g) L. Bernardi and K. A. Jørgensen, Chem. Commun., 2005, 1324 RSC; (h) Y. Hamashima, T. Suzuki, Y. Shimura, T. Himizu, N. Umebayashi, T. Tamura, N. Sasamoto and M. Sodeoka, Tetrahedron Lett., 2005, 46, 1447 CrossRef CAS; (i) N. Shibata, J. Kohno, K. Takai, T. Ishimura, S. Nakamura, T. Toru and S. Kanamasa, Angew. Chem., Int. Ed., 2005, 44, 4204 CrossRef CAS; (j) Y. Hamashima, T. Suzuki, H. Takano, Y. Shimura and M. Sodeoka, J. Am. Chem. Soc., 2005, 127, 10164 CrossRef CAS.
- D. Y. Kim and E. J. Park, Org. Lett., 2002, 4, 545 CrossRef CAS.
- See e.g.: (a) S. France, A. Weatherwax and T. Lectka, Eur. J. Org. Chem., 2005, 475 CrossRef CAS; (b) S. France, H. Wack, A. E. Taggi, A. M. Hafez, T. R. Wagerle, M. H. Shah, C. L. Duisch and T. Lectka, J. Am. Chem. Soc., 2004, 126, 4245 CrossRef CAS.
- M. P. Brochu, S. P. Brown and D. W. C. MacMillan, J. Am. Chem. Soc., 2004, 126, 4108 CrossRef CAS.
- N. Halland, A. Braunton, S. Bachmann, M. Marigo and K. A. Jørgensen, J. Am. Chem. Soc., 2004, 126, 4790 CrossRef CAS.
- M. Marigo, S. Bachmann, N. Halland, A. Braunton and K. A. Jørgensen, Angew. Chem., Int. Ed., 2004, 43, 5507 CrossRef.
- (a) D. Enders and R. M. Hüttl, Synlett, 2005, 991 CrossRef CAS; (b) M. Marigo, D. Fielenbach, A. Braunton, A. Kjærsgard and K. A. Jørgensen, Angew. Chem., Int. Ed., 2005, 44, 3703 CrossRef CAS; (c) D. D. Steiner, N. Mase and C. F. Barbas III, Angew. Chem., Int. Ed., 2005, 44, 3706 CrossRef CAS; (d) T. D. Beeson and D. W. C. MacMillan, J. Am. Chem. Soc., 2005, 127, 8826 CrossRef CAS.
- K. Omura, J. Org. Chem., 1996, 61, 2006 CrossRef CAS.
- P. L. Bailey, A. D. Briggs, R. F. W. Jackson and J. Pietrizka, J. Chem. Soc., Perkin Trans. 1, 1998, 3359 RSC.
- D. Rogers, Acta Crystallogr., Sect. A, 1981, A37, 734 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Full experimental details. See http://dx.doi.org/10.1039/b509366j |
| ‡ CCDC 277076. Crystals of 4i, 3-bromo-tetrahydro-pyran-4-one, C5H7BrO2, M = 179.01, are orthorhombic, P212121, unit cell: a = 4.3135(4), b = 11.327(1), c = 12.558(1) Å, V = 613.57(9) Å3, Z = 4, μ(Mo-Kα) = 6.602 mm−1. A total of 15870 reflections (2615 independent) were collected at 100 K on an APEX diffractometer with CCD detector. The structure was solved by direct methods and refined by least squares on F to a final R = 0.031, Rw = 0.033, GOF = 0.870 using 2585 reflections with I > 0.103 parameters refined. A parameter according to Rogers12 refined against all positive reflections including 1063 Bijvoet pairs gave the value 1.00(2) establishing the (R) configuration at C3. See http://dx.doi.org/10.1039/b509366j for crystallographic data in CIF or other electronic format. |
| This journal is © The Royal Society of Chemistry 2005 |