Combining two-directional synthesis and tandem reactions: desymmetrisation by intramolecular cycloaddition/triazoline fragmentation†‡
Martin
Rejzek
a,
Robert A.
Stockman
*a,
Jan H.
van Maarseveen
b and
David L.
Hughes
c
aSchool of Chemical Sciences and Pharmacy, University of East Anglia, Norwich, UK NR4 7TJ. E-mail: r.stockman@uea.ac.uk; Fax: 44 (0)1603 592005; Tel: +44 (0) 1603 593890
bInstitute of Molecular Chemistry, University of Amsterdam, Niewe Achteragracht 129, 1018 WS Amsterdam, The Netherlands. E-mail: jvm@science.uva.nl; Fax: (+31)
(0) 20 5255670; Tel: (+31)
(0)20 5255671
cX-Ray Crystallography Centre, School of Chemical Sciences and Pharmacy, University of East Anglia, Norwich, UK NR4 7TJ
First published on 31st August 2005
Abstract
A tandem azide formation/intramolecular cycloaddition/triazoline fragmentation/Michael addition, which results in a non-symmetrical quinolizidine from an acyclic symmetrical precursor, is presented.
The strategies of two-directional synthesis1 and tandem reactions2 both offer the potential to substantially reduce the number of operations required to synthesise complex molecules. As part of an on-going programme studying the potential of combining two-directional synthesis and tandem reactions,3 we herein disclose a remarkable self-desymmetrising cascade reaction.
Two-directional synthesis intrinsically generates symmetrical products. By using two-directional synthesis as a means of forming simple symmetrical functionalised chains, then using a tandem reaction to “fold” these chains, creating rings and stereocentres, we have the potential to form complex polycyclic compounds in just a few steps. Our recent syntheses of perhydrohistrionicotoxin4 and hippodamine5 exemplify this approach to the synthesis of complex alkaloids.
During an investigation related to our work on hippodamine, we wished to synthesise symmetrical azide 3, such that we could investigate a Staudinger-type azide reduction/tandem Michael cyclisation. In order to access azide 3, we started with the symmetrical ketone 1 (Scheme 1), which is available in 4 steps from 1,3-dithiane, or in 5 steps from ethyl formate.6 Reduction of the ketone with sodium borohydride followed by conversion of the resulting pseudo-C2 symmetric alcohol to mesylate 2 proceeded uneventfully in 96% yield over two steps. Reaction of mesylate 2 with sodium azide in DMF at 50 °C was expected to result in the formation of azide 3. TLC showed one clear product, with a further amount of baseline material, which we considered was possibly due to azide overaddition/decomposition. However, upon inspection of the crude 1H NMR, it was quickly obvious that something far more complicated than azide 3 had been formed in the reaction pot. Indeed, after purification by column chromatography, it was found that the sole isolable product of the reaction was quinolizidine derivative 5 (52%). Key observations which led us to assign the structure of bicyclic diazo compound 5 were the obvious asymmetry present in the 1H and 13C spectra (including two ester carbonyl peaks in the 13C NMR spectrum), and the characteristic diazo stretch in the IR at 2150 cm−1. High resolution mass-spectral data confirmed the molecular mass of 338.2075 (M + H). However, in order to further confirm the stereochemistry of our product, we decided it would be best to make a crystalline derivative suitable for X-ray structure determination. Thus upon exposure to standard hydrogenation conditions (10% Pd/C, 1 atm hydrogen), diazo compound 5 was quantitatively converted to the crystalline hydrazone 6, whose X-ray structure7 is shown in Fig. 1.
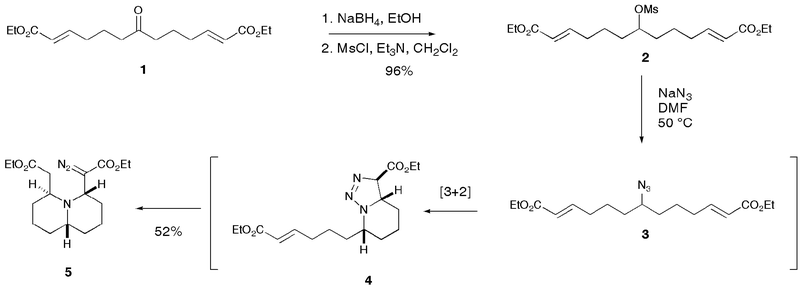 | ||
| Scheme 1 Two-directional synthesis and tandem reaction of azide 3. | ||
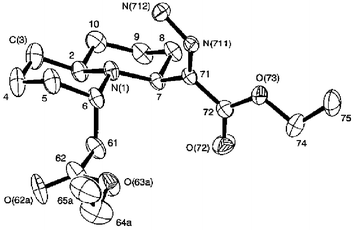 | ||
| Fig. 1 X-ray structure of hydrazone 6, formed by hydrogenation of 5. | ||
Study of the X-ray structure of 6, combined with the isolation of a small amount of triazoline 4 from the reaction mixture when stopped early, has allowed us to postulate a mechanism for this tandem reaction sequence, which is presented in Scheme 2. Thus, [3 + 2] cycloaddition of azide 3 with one of the enoate functionalities results in the thermodynamically most favoured cycloadduct 4. 1,4-Prototopic shift within the triazoline of 4, followed by fragmentation to yield diazo 2,6-disubstituted piperidine 7 and subsequent Michael-type ring closure is the most satisfactory mechanism. Although the triazoline fragmentation8 to a piperidine is precedented under basic conditions,9 lending weight to this mechanistic proposal, we were unable to detect quantities of the intermediate piperidine 7. However, we were able to detect these types of piperidine intermediate in our hippodamine synthesis,5 which used a similar Michael-type ring closure to a 4,6-disubstituted quinolizidine. It could also be envisaged that the formation of 5 from 4 could be achieved through a concerted ene-type mechanism, with the tertiary nitrogen and the proton α to the ester on the triazoline being delivered to either end of the electron deficient alkene, with concomitant triazoline fragmentation.
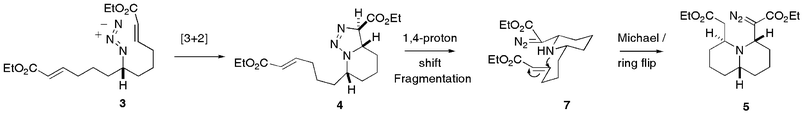 | ||
| Scheme 2 Proposed mechanism for the formation of 5. | ||
In conclusion, the power of combining two-directional synthesis and tandem reactions has provided a short and efficient entry into a non-symmetrical 4,6-disubstituted quinolizidine skeleton. Further studies into the use of the tandem azide formation/[3 + 2]/fragmentation reaction are on-going and our results will be reported in due course.
The authors wish to thank the Leverhulme Trust for funding and the EPSRC Mass Spectrometry Service, Swansea.
Notes and references
- S. R. Magnuson, Tetrahedron, 1995, 51, 2167 CrossRef CAS; C. S. Poss and S. L. Schreiber, Acc. Chem. Res., 1994, 27, 9 CrossRef CAS.
- R. A. Bunce, Tetrahedron, 1995, 51, 13103 CrossRef CAS.
- For the previous paper in this series (Part 6), see: P. McDermott and R. A. Stockman, Org. Lett., 2005, 6, 27 Search PubMed.
- R. A. Stockman, A. Sinclair, L. G. Arini, P. Szeto and D. L. Hughes, J. Org. Chem., 2004, 69, 1598 CrossRef CAS.
- M. Rejzek and R. A. Stockman, Org. Biomol. Chem., 2005, 3, 73 RSC.
- L. G. Arini, P. Szeto, D. L. Hughes and R. A. Stockman, Tetrahedron Lett., 2004, 45, 8371 CrossRef CAS.
- Crystal data for 6: C17H29N3O4, M = 339.4. Triclinic, space group P-1, a = 8.048(2), b = 15.296(3), c = 7.872(3) Å, α = 104.54(2), β = 93.23(3), γ = 80.36(2)°, V = 924.7(4) Å3. Z = 2, Dc = 1.219 g cm−3, F(000) = 368, T = 293(1) K, μ(Mo-Kα) = 0.9 cm−1, λ(Mo-Kα) = 0.71069 Å. Crystals are clear, colourless plates. Intensity data were measured on a Rigaku/MSC AFC7R diffractometer (Mo-Kα radiation, graphite monochromator); 3533 reflections (θmax = 25°), 3277 unique (Rint = 0.034), 2006 ‘observed’ with I > 2σI. Structure determined by direct methods (SHELXS-97 program10a); refinement by full-matrix least-squares methods, on F2's, in SHELXL-97.10b One ester group is disordered equally in two alternative orientations. Two hydrogen atoms were located on the β-nitrogen atom of the NNH2 group and refined freely; all other hydrogen atoms were included in idealised positions with Uiso values ‘riding’. At convergence, wR2 = 0.166 and R1 = 0.108 for all 3277 reflections weighted w = [σ2(Fo2) + (0.0790P)2]−1 with P = (Fo2 + 2Fc2)/3; for ‘observed’ data only, R1 = 0.060. CCDC 277408. See http://dx.doi.org/10.1039/b508969g for crystallographic data in CIF or other electronic format.
- R. Huisgen, G. Szeimies and L. Möbius, Chem. Ber., 1966, 99, 475 CrossRef CAS.
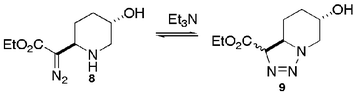
- C. Herdeis and T. Schiffer, Tetrahedron, 1996, 52, 14745 CrossRef CAS; T. Herdeis and T. Schiffer, Synthesis, 1997, 1405 CrossRef
Base induced equilibration of triazolidine and diazoester was found to be facile by Herdeis. - (a) G. M. Sheldrick, SHELXS-97, Program for solution of crystal structures, University of Göttingen, Germany, 1997 Search PubMed; (b) G. M. Sheldrick, SHELXL-97, Program for refinement of crystal structures, University of Göttingen, Germany, 1997 Search PubMed.
Footnotes |
| † Part 7 in our series concerning two-directional synthesis and tandem reactions. |
| ‡ This paper is dedicated to the memory of Mark Jones. |
| This journal is © The Royal Society of Chemistry 2005 |