Nanoscale Borromean links for real†
Andrea J.
Peters
,
Kelly S.
Chichak
,
Stuart J.
Cantrill
and
J. Fraser
Stoddart
California NanoSystems Institute and Department of Chemistry and Biochemistry, University of California, Los Angeles, 405 Hilgard Avenue, Los Angeles, California 90095, USA. E-mail: stoddart@chem.ucla.edu; Fax: +1 310 206 1843; Tel: +1 310 206 7078
First published on 10th June 2005
Abstract
Borohydride reduction of a Borromean Ring (BR) complex containing six zinc(II) ions and 12 imine bonds has resulted in its demetallation, producing a neutral BR compound and also its free macrocycle, following cleavage of at least one of the imine bonds in the ethanolic reaction mixture.
The synthesis of topologically distinctive molecular compounds,1 which has taxed the intellects and tested the dexterity of chemists for some time now, has led to the emergence recently2,3 of nanoscale Borromean Rings (BRs) in addition to the much more common molecular catenanes and knots.4 The production of the nanoscale BRs has been accomplished using an approach in which a collection of molecular recognition elements, including six metal–ligand coordination sites and six donor–acceptor–donor interactive units, act in unison with supreme efficiency.
The template-directed synthesis5 of the nanoscale BRs (Fig. 1a,b) relies upon (i) the dynamic covalent chemistry6 associated with the formation of 12 imine bonds7 from six 2,6-diformylpyridine molecules and six diaminobipyridine ligands, (ii) the dynamic coordination chemistry8 involving six exo-oriented bipyridine ligands and six endo-oriented tridentate bis-Schiff bases in the formation of 30 dative bonds and (iii) the dynamic supramolecular chemistry9 surrounding the stabilisation afforded by 12 donor–acceptor interactions between pairs of para-disubstituted phenolic residues and 2,2′-bipyridine ligands chelated to six zinc(II) ions. Even when the nanoscale BRs have reached molecular fruition, there is still the opportunity for them to exchange their 2,6-diformylpyridine components under the appropriate conditions. Although we have recognised10 this scrambling ability, following experiments where the pyridine diimine precursors are labelled with chlorine and bromine atoms in their 4-positions, we believe the overall structural integrity of the molecules is largely maintained during the exchange process. Bearing in mind that one of the fundamental properties of the Borromean link topology is the fact that, as soon as one of the three rings is severed, the other two rings part company, no evidence was found at all in the scrambling experiments10 for this level of disassembly. It is clear that, in order to produce real Borromean links (Fig. 1c) on a molecular scale from the BR compounds already described in the literature,2,10 we have to achieve two goals simultaneously – one is to remove all six zinc(II) ions from the molecule and the other is to reduce all 12 imine bonds in the molecules to secondary amine functions. In this communication, we report the realisation of the fully demetallated‡ molecular Borromean links, and also provide a chemical proof§ of their structure, i.e., when one of the three rings is cleaved, the other two can fall apart (Scheme 1).
 | ||
| Fig. 1 (a) Crystal structure of the BR complex 1. Graphical representations of (b) the Zn(II)-containing complex 1 and (c) the metal-free BR compound 2. The red, blue and green macrocycles have identical constitutions. The thinner regions of the tubular rings in (b) represent the dynamic imine bonds present in 1. | ||
 | ||
| Scheme 1 Graphical representation of the cleavage of one of the macrocycles (blue) during the borohydride reduction reaction of 1 resulting in the disassembly of the BRs to produce two macrocycles, which fall apart spontaneously to give 3 and a linear component 4. | ||
The production of the demetallated BRs 2 was accomplished (Scheme 2; Conditions A) by treating a 1.4 mM ethanolic solution of 1 with an excess of NaBH4 at 22 °C in an inert atmosphere. After stirring the solution at this temperature for 5 days, the reaction mixture was quenched with H2O, followed by the addition of an excess of EDTA in order to remove the Zn(II) ions. After work-up of the reaction and isolation of the product (see Electronic Supplementary Information†), characterisation was achieved by high resolution electrospray ionisation (HR-ESI) mass spectrometry and 1H NMR spectroscopy. A preliminary 1H NMR spectroscopic investigation in CD3SOCD3 of the crude product isolated from the reaction mixture revealed (Fig. 2b) the absence of the peak for imine protons which were present (Fig. 2a) as a singlet (δ = 9.02 ppm) in the 1H NMR spectrum of 1. Analysis of the spectrum (Fig. 2b) revealed, however, that there were two sets of seven resonances, indicating that more than one highly symmetrical product had been formed¶ during the borohydride reduction. Although the corresponding peaks in each of the two sets displayed the same coupling constants, one set of peaks was shifted slightly upfield with respect to the other set. By comparing the 1H NMR spectra shown in Figs. 2a and 2b, one of the products of the reaction can be assigned to one set of seven signals centred on δ = 6.36 (H-g), 6.59/7.01 (H-e/f), 7.21 (H-b), 7.46 (H-i), 7.62 (H-a) and 7.89 (H-h) ppm, while the other product can be associated with another set of seven signals centred on δ = 6.87 (H-g′), 7.04/7.41 (H-e′/f′), 7.32 (H-b′), 7.64 (H-i′), 7.74 (H-a′) and 8.34 (H-h′) ppm. In a second demetallation in which a 1.0 mM ethanolic solution of 1 was heated with an excess of NaBH4 at 80 °C for 16 h in an inert atmosphere (Scheme 2; Conditions B), the 1H NMR spectrum, which was recorded after work-up of the reaction, revealed (Fig. 2c) the presence of only one of the two sets of seven signals – namely, the downfield set – identified in the product of the first demetallation.
 | ||
| Fig. 2 Partial 1H NMR spectra (600 MHz) recorded in CD3SOCD3 of (a) the BR complex 1 and the contents of reaction mixtures containing (b) the fully reduced, demetallated BR compound 2 plus free macrocycle 3 and (c) the free macrocycle 3. | ||
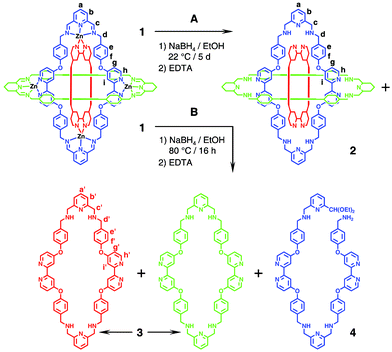 | ||
| Scheme 2 Schematic representation of 1 and 2, in which only the blue ring depicts all of the atoms and bonds present in the macrocycles that make up the BR structure. Reduction of the imine bonds in 1 followed by demetallation of the BR complex in an ethanolic solution (A) at 22 °C, produces the demetallated BR 2 along with 3 and 4, and (B) at 80 °C, only the macrocycle 3 and linear fragment 4. | ||
Further investigation by HR-ESI mass spectrometry helped to reveal the identity of the two products: addition of one drop of TFA to the first reaction mixture (Conditions A), prior to recording its HR-ESI-MS, revealed (Fig. 3a) the presence of three major peaks at m/z of 1505.6834, 1004.1120, and 753.3363, corresponding to [M + 2H]2+, [M + 3H]3+ and [M + 4H]4+ respectively, an observation which is consistent with the product being the fully reduced, completely demetallated BRs, i.e., 2. The HR-ESI-MS of the product from the second demetallation (Conditions B), treated likewise with a drop of TFA, revealed (Fig. 3b) one major singly-charged peak for [M + H]+ at 1003.4418, an m/z value which correlates exactly with the mass of only one ring fully reduced, viz., 3, for which the calculated [M + H]+ is 1003.4402, in keeping with the molecular formula, C62H54N10O4. The result indicates that, in the second demetallation, where the borohydride reduction was carried out at an elevated temperature (80 °C), the three rings have indeed fallen apart during the reaction. We suggest that, under basic conditions, the imines can react with trace amounts of ethoxide ions to form a tetrahedral O-ethyl hemiaminal intermediate that produces, after work-up of the reaction, the free aldehyde from the hydrated one or the diethyl acetal 4.|| The result of the irreversible scission of one of the rings allows, once the demetallation reaches completion, the other two rings to fall apart and hence to be isolated as their fully reduced derivatives, i.e., 3, along with a linear fragment which we suggest could be the diethyl acetal 4. There is evidence from the HR-ESI-MS of the TFA-treated ethanolic solution of the reaction mixture for the presence of the linear fragment containing the diethyl acetal 4 (see Electronic Supplementary Information†). A singly charged peak with a m/z of 1093.5147 for [M + H]+ equates with a calculated [M + H]+ of 1093.5083 for M = C66H64N10O6. Although it is possible that other linear fragments are formed during the borohydride reduction, there is no evidence for them in the mass spectrum.
 | ||
| Fig. 3 HR-ESI-MS of reaction mixtures (a) A in methanol plus TFA showing the presence of peaks corresponding to the reduced BRs 2 and (b) B in ethanol plus TFA showing the presence of peaks corresponding to the macrocycle 3 and linear fragment 4. | ||
The information obtained from mass spectrometry provides further insight into the 1H NMR spectroscopic data (Fig. 2b) acquired from the first (Conditions A) of the two borohydride reductions performed on 1. It indicated the presence of two highly symmetrical products, namely the fully reduced, demetallated BRs 2 and the component macrocycle 3 in which all four imine bonds have been reduced to secondary amine functions. In Fig. 2b, we attribute (see Electronic Supplementary Information†) the upfield set of seven signals to 2 and the downfield set to 3. The fact that the resonances for 2 appear at higher field than those for 3 suggests that there are π–π stacking interactions present in the BR compound that are absent in its ring components when they become free from each other.
Aside from this piece of research providing the intellectually satisfying chemical proof§ for the Borromean link topology in 1, it has produced a real BR compound, minus its metal ion templates, where the three rings are genuinely mechanically interlocked according to the expected topology.
This work was supported in part by a National Science Foundation (NSF) grant (CHE0317170) and two NSF equipment grants (CHE9974928 and CHE0092036).
Notes and references
- (a) H. L. Frisch and E. Wasserman, J. Am. Chem. Soc., 1961, 83, 3789–3795 CrossRef CAS; (b) S. J. Tauber, J. Res. Natl. Bur. Stand. A, 1963, 67, 591–599 Search PubMed; (c) V. I. Sokolov, Russ. Chem. Rev., 1973, 43, 452–463 Search PubMed; (d) D. M. Walba, Tetrahedron, 1985, 41, 3161–3212 CrossRef CAS; (e) J. Simon, Proc. Symp. Appl. Math., 1992, 45, 97–130 Search PubMed; (f) N. van Gulick, New J. Chem., 1993, 17, 619–625 Search PubMed; (g) C. Liang and K. Mislow, J. Math. Chem., 1994, 16, 27–35 CrossRef CAS; (h) K. Mislow, Top. Stereochem., 1999, 22, 1–82 Search PubMed; (i) J. C. Loren, M. Yoshizawa, R. F. Haldimann, A. Linden and J. S. Siegel, Angew. Chem., Int. Ed., 2003, 42, 5702–5705 CrossRef CAS.
- K. S. Chichak, S. J. Cantrill, A. R. Pease, S.-H. Chiu, G. W. V. Cave, J. L. Atwood and J. F. Stoddart, Science, 2004, 304, 1308–1312 CrossRef CAS.
- (a) J. S. Siegel, Science, 2004, 304, 1256–1258 CrossRef CAS; (b) C. A. Schalley, Angew. Chem., Int. Ed., 2004, 43, 4399–4401 CrossRef CAS; (c) S. J. Cantrill, K. S. Chichak, A. J. Peters and J. F. Stoddart, Acc. Chem. Res., 2005, 38, 1–9 CrossRef CAS.
- J.-P. Sauvage and C. Dietrich-Buchecker, eds., Molecular Catenanes, Rotaxanes and Knots: A Journey Through the World of Molecular Topology; Wiley-VCH: Weinheim, Germany, 1999 Search PubMed.
- F. Diederich and P. J. Stang, eds., Templated Organic Synthesis; Wiley-VCH: Weinheim, Germany, 2000 Search PubMed.
- (a) J.-M. Lehn and A. V. Eliseev, Science, 2001, 291, 2331–2332 CrossRef CAS; (b) O. Ramström and J.-M. Lehn, Nat. Rev. Drug Discov., 2002, 1, 26–36 CrossRef CAS; (c) S. J. Rowan, S. J. Cantrill, G. R. L. Cousins, J. K. M. Sanders and J. F. Stoddart, Angew. Chem., Int. Ed., 2002, 41, 898–952 CrossRef; (d) B. Fuchs, A. Nelson, A. Star, J. F. Stoddart and S. B. Vidal, Angew. Chem., Int. Ed., 2003, 42, 4220–4224 CrossRef CAS; (e) R. T. S. Lam, A. Belengaer, S. L. Roberts, C. Naumann, T. Jarrosson, S. Otto and J. K. M. Sanders, Science, 2005, 308, 667–669 CrossRef CAS.
- For stoppering approaches to rotaxanes incorporating imine-containing dumbbells, see (a) S. J. Cantrill, S. J. Rowan and J. F. Stoddart, Org. Lett., 1999, 1, 1363–1366 CrossRef CAS; (b) S. J. Rowan and J. F. Stoddart, Org. Lett., 1999, 1, 1913–1916 CrossRef CAS; for clipping approaches to rotaxanes incorporating imine-containing macrocycles, see (c) P. T. Glink, A. I. Oliva, J. F. Stoddart, A. J. P. White and D. J. Williams, Angew. Chem., Int. Ed., 2001, 40, 1870–1875 CrossRef CAS; (d) M. Horn, J. Ihringer, P. T. Glink and J. F. Stoddart, Chem. Eur. J., 2003, 9, 4046–4054 CrossRef CAS; (e) L. Hogg, D. A. Leigh, P. J. Lusby, A. Morelli, S. Parsons and J. K. Y. Wong, Angew. Chem., Int. Ed., 2004, 43, 1218–1221 CAS; (f) F. Arico, T. Chang, S. J. Cantrill, S. I. Khan and J. F. Stoddart, Chem. Eur. J., 2005, 11 DOI:10.1002/chem.200500148.
- (a) M. Fujita, Acc. Chem. Res., 1999, 32, 53–61 CrossRef CAS; (b) S. Leininger, B. Olenyuk and P. J. Stang, Chem. Rev., 2000, 100, 853–908 CrossRef CAS; (c) D. A. Leigh, P. J. Lusby, S. J. Teat, A. J. Wilson and J. K. Y. Wong, Angew. Chem., Int. Ed., 2001, 40, 1538–1543 CrossRef CAS; (d) M.-J. Blanco, J.-C. Chambron, M. C. Jiménez and J.-P. Sauvage, Top. Stereochem., 2003, 23, 125–173 Search PubMed.
- (a) J.-M. Lehn, Supramolecular Chemistry, VCH: Weinheim, Germany, 1995 Search PubMed; (b) M. C. T. Fyfe and J. F. Stoddart, Acc. Chem. Res., 1997, 30, 393–401 CrossRef CAS; (c) J.-M. Lehn, Science, 2002, 295, 2400–2403 CrossRef CAS; (d) D. N. Reinhoudt and M. Crego-Calama, Science, 2002, 295, 2403–2407 CrossRef CAS.
- K. S. Chichak, S. J. Cantrill and J. F. Stoddart, Chem. Commun., 2005 10.1039/b503717d.
Footnotes |
| † Electronic supplementary information (ESI) available: synthesis, 1H–1H COSY, MS, ion-binding. See http://www.rsc.org/suppdata/cc/b5/b505730b/index.sht |
| ‡ As a general descriptor for molecules possessing this distinctive topology, it seems that ‘Borromean ring compounds’ is an accurate and accepted form of nomenclature. We propose, however, that Borromean ring compounds which are assembled using metal ions, and retain those metal ions as an intrinsic part of their structure, be referred to as ‘Borromeates’. Furthermore, upon removal of the metal ions, the term ‘Borromeand’ is suggested for the resulting metal-free compound. This terminology is akin to that introduced by Sauvage to deal more accurately with the descriptions of metal ion-containing catenanes and their derivatives – which he refers to as ‘catenates’, that become ‘catenands’ upon demetallation. It should be noted, however, that, in the case of the Borromeate presented in this communication, the Borromeand is not obtained through simple demetallation alone, but upon demetallation and reduction of the imine bonds. The term ‘catenand’ was introduced in 1984. See: C. O. Dietrich-Buckecker, J.-P. Sauvage and J.-M. Kern, J. Am. Chem. Soc., 1984, 106, 3043–3045. The term ‘catenate’ followed soon thereafter. See: M. Cesario, C. O. Dietrich-Buchecker, J. Guilhem and J.-P. Sauvage, J. Chem. Soc., Chem. Commun., 1985, 244–247. The ‘catenate/catenand’ terminology was an extension of Lehn's earlier ‘cryptate/cryptand’ terminology. See: J.-M. Lehn, Angew. Chem., Int. Ed. Engl., 1988, 27, 89–112. |
| § The mixture of products obtained in this experiment is tantamount to a proof of the Borromean link topology of the starting material, insofar as no catenated (or otherwise interlocked) compounds are observed – only the macrocycle 3 and its linear progeny 4 are formed. Such a degradative proof of this particular molecular topology was first employed by Seeman in the characterisation of single-stranded DNA-based Borromean rings. The incorporation of a different restriction site in each individual circuit of this hetero-Borromean ensemble allowed for the separate and targeted cleavage of each ring upon treatment with a specific endonuclease. Indeed, in each case, this enzymatic scission yielded a mixture of products consistent with the Borromean link topology – namely, two intact macrocycles, as well as the linear debris resulting from the destruction of the third ring in the original structure. See: C. D. Mao, W. Q. Sun and N. C. Seeman, Nature, 1997, 386, 137–138. |
| ¶ Attempts to purify the reaction mixture using column chromatography and recrystallization were unsuccessful. This failure arose from the fact that the compounds adhere to both silica and alumina stationary phases on TLC in addition to the extreme insolubility of the compounds in common organic solvents, respectively. |
| || Under basic conditions, the rate of hydrolysis of the chelated Schiff base will be independent of pH and the rate determining step will be the nucleophilic attack of ethoxide ions on the chelated Schiff base. The hydrolysis of a Schiff base in aqueous solutions normally proceeds through a pH-dependent zwitterionic carbinolamine species, but, for the BRs this intermediate cannot be formed in ethanolic solution and hydrolysis of the imine to the aldehyde probably does not occur. Instead the Lewis acidic nature of zinc(II) ions could catalyse the nucleophilic attack of the Schiff base by ethoxide ions, producing a tetrahedral O-ethyl hemiaminal intermediate. If the amine is expelled from this ethanol adduct of the tetrahedral aminal then the diethyl acetal will more than likely be produced by solvolysis of the ethyloxonium intermediate. The diethyl acetal will be inert towards reduction and should be stable under basic condition. See E. H. Cordes and W. P. Jencks, J. Am. Chem. Soc., 1963, 85, 2843–2848. |
| This journal is © The Royal Society of Chemistry 2005 |