Catalytic molecular motors: fuelling autonomous movement by a surface bound synthetic manganese catalase†
Javier
Vicario
,
Rienk
Eelkema
,
Wesley R.
Browne
,
Auke
Meetsma
,
René M.
La Crois
and
Ben L.
Feringa
*
Department of Organic and Molecular Inorganic Chemistry, Stratingh Institute, University of Groningen, Nijenborgh 4, 9747 AG, Groningen, The Netherlands. E-mail: B.L.Feringa@rug.nl
First published on 31st May 2005
Abstract
A molecular approach to the powering of multi-component nano-devices capable of autonomous translational and rotational motion through the conversion of chemical to kinetic energy is reported.
The effective use of nano- and microscale machinery capable of driving linear and rotary motion is essential to the proper functioning of living cells.1 The construction of artificial molecular machines, ubiquitous in nature, remains, however, a major contemporary challenge.2 The recent development of molecular based systems including shuttles,3 rotors,4 muscles,5 switches,6 elevators,7 motors8 and processive catalysts9 has brought the prospect of synthetic molecular based ‘mechanical machines’ within sight. Nevertheless the required ‘fuelling’ of the motion of these molecular systems remains a major hurdle. Indeed, central to the function of nanoscopic (e.g. ATP-synthase, which employs H+ gradients) and microscopic (e.g. ATP-driven actin filaments in muscle tissue, controlled by Ca2+ gradients) molecular devices employed by nature is the ability to convert chemical to mechanical energy.1,10 Although light-powered molecular devices, such as the first molecular motor capable of performing repetitive unidirectional rotary motion, developed in our group,8a,11 hold considerable potential towards driving changes in wholly molecular systems, as demonstrated recently in liquid crystal films,12 the movement of larger assemblies (i.e. 10−4–10−9 m) requires concerted action of several molecular motors or significantly greater power to effect rotational and translational motion. The high power density required might be achievable through the use of chemical energy. Indeed Kelly and coworkers8b have demonstrated that rotary motion can be induced by sequential chemical conversions. An attractive alternative to the application of complex chemical transformations to drive molecular based devices is provided by the approach taken recently by the groups of Whitesides,13 Sen, Mallouk and Crespi,14 and Ozin and Manners,15 in which the decomposition of H2O2 on the surface of microscopic composite (bi-)metallic objects is employed to achieve translational and/or rotational movement through the creation of localised [O2] gradients. The nature of the motion induced by this approach was found to be critically dependent on both the particle dimensions and the ability to localise the production of O2. The use of a synthetic oxygen evolving complex might be of considerable advantage to future (non-metallic) micro- and nanoscale molecular machines, as it can be easily (chemically) modified, tether length can be adjusted, and local catalyst density is controllable. Furthermore, it offers the possibility to control oxygen evolution down at the molecular level. A synthetic molecular system capable of chemically driven motion still remains a major hurdle, however, due to the requirement to be able to immobilise molecular catalysts on particles with retention of catalyst function. Furthermore the integrity of the multicomponent system created must not be compromised during the conversion of chemical to kinetic energy.
Here, we report our preliminary results on autonomous movement of microparticles (μ-particles) via a [covalently] tethered synthetic catalase mimic. The design (Fig. 1) of a molecular based device capable of converting chemical energy to kinetic energy, comprises three key elements: an object (micro- or nanoparticle, complex or macro-molecule) to be transported, a spacer/tether and a catalyst for converting chemical energy to kinetic energy. The new highly efficient catalase mimic, [(Mn(II)(L−))2(RCO2−)]+ (1) (where HL is 2-{[[di(2-pyridyl)methyl](methyl)amino]methyl}phenol) developed within our group, is well suited to such a role. Its high rate of disproportionation of H2O2 to H2O and O2 (i.e. catalase activity) and the ability to attach the catalyst covalently, through either the ligand L− or more promisingly the bridging carboxylate group, allows for facile fixation to the surface of nano- and micro-scale objects through chemical modification.
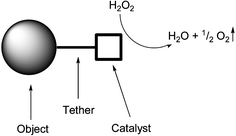 | ||
| Fig. 1 Design of a system propelled by a catalase mimic. | ||
Designed as a functional model for dinuclear Mn-catalase enzymes,16 ligand HL, bearing a phenol, tertiary amine and two pyridine binding sites, was synthesised. Complexation with Mn(ClO4)2 in the presence of carboxylic acids provided dinuclear MnIIMnII-complexes 1a and 1b (Scheme 1). X-ray analysis of the benzoate complex 1a confirms the bridging nature of the phenolates and the carboxylate (Fig. 2, see also ESI†).17 The ability of 1 to catalyse H2O2 disproportionation was demonstrated in several solvents18 with turn over frequencies (T.O.F.) of ∼0.27 s−1 at 25 °C (i.e. CH3CN, 30% aq. H2O2).
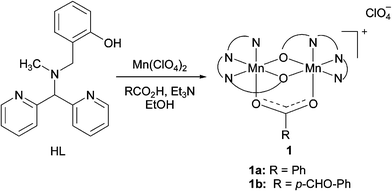 | ||
| Scheme 1 | ||
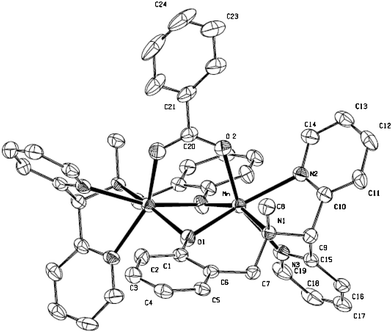 | ||
| Fig. 2 X-ray structure of 1a. | ||
Covalent immobilisation of the catalyst via the bridging carboxylate ligand was achieved using the p-formyl-functionalised complex 1b, which was attached to aminopropyl-modified silica microparticles (40–80 μm) in ethanol–acetonitrile through imine formation (Scheme 2). Multiple rigorous washing steps (EtOH, CH3CN, CH2Cl2 and Et2O) of the μ-particles were performed to remove residual, non-covalently bound, complex 1b. The modification of the μ-particles via an imine bridge was confirmed by IR spectroscopy and electrochemistry (see ESI†), through comparison of the aminopropyl-functionalised particles, its iminobenzoic acid analogue, and the particles functionalised with 1b as well as free complex. The immobilised catalytic system obtained was found to be robust to detachment (i.e. retains activity, IR and redox properties are unaffected, see ESI†) from the μ-particles and exhibited high activity towards H2O2 disproportionation as observed with the free catalyst (e.g.1a). The modified particles were recoverable by filtration and could be recycled several times without significant loss in activity. The characteristic imine absorption at 1645 cm−1 is not affected by H2O2 decomposition and IR spectra of functionalised μ-particles are unaffected by catalase activity. Kinetic and thermodynamic parameters of H2O2 decomposition by functionalised μ-particles as well as free complex 1b (5 wt%) were obtained by Eyring plot analysis,19 showing first order kinetics (w.r.t. [H2O2]) with large negative entropy of activation (k° = 3.09 × 10−3 s−1, ΔS‡ = −288.62 J K−1mol−1 for the free complex 1b and k° = 1.51 × 10−3 s−1, ΔS‡ = −291.07 J K−1mol−1 for the 1b-functionalised microparticles).
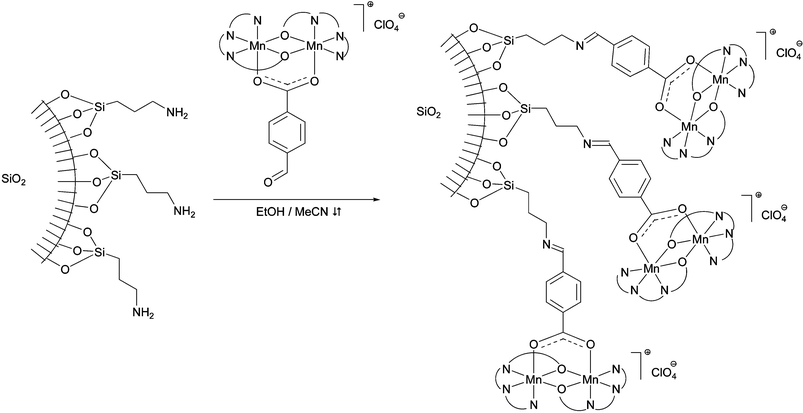 | ||
| Scheme 2 | ||
In order to study the movement induced by O2 formation, a thin liquid film, containing a dilute suspension (in CH3CN or glycerol) of the 1b-functionalised μ-particles, mixed with non-functionalised μ-particles (40–80 μm) as reference, on a microscope glass slide, were monitored after addition of a 5% H2O2 solution in CH3CN. O2 evolution was observed as small O2 bubbles appearing on only the catalyst-functionalised μ-particles. Both linear and rotary motion of the modified μ-particles was found. The translational motion of a μ-particle (see movie in ESI†) and the trajectory followed until the supply of H2O2 is insufficient to sustain movement, is seen in Fig. 3. The measured average speed for this particle is 35 μm s−1.
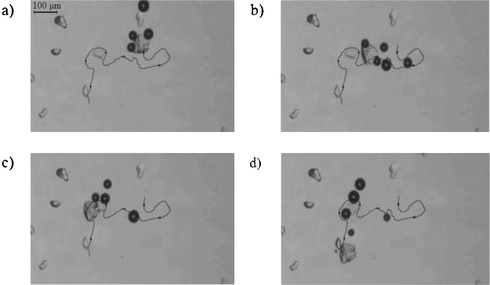 | ||
| Fig. 3 Pathway of an 80 μm 1b-functionalised SiO2 particle in CH3CN and its position at a) 0 s, b) 4 s, c) 14 s and d) 18 s (see supplementary movie†). | ||
As the dinuclear Mn-complex 1b is a highly active catalyst for H2O2 decomposition large disturbance of the dispersed film and numerous collisions of μ-particles are observed at higher H2O2 concentrations. The direction of the movement is in accordance with that of the system of Whitesides and coworkers, with O2 evolution at the trailing end of the moving objects.13 The non-symmetric nature of the particle with respect to defects capable of causing cavitation and hence bubble formation rather than inhomogeneous distribution of catalyst is likely to be the dominant factor in determining the directionality of the motion induced. Interestingly, the autonomous moving μ-particles also pass along static particles (Fig. 3b,c) on the surface, changing directionality.
Slow rotational movement of a functionalised μ-particle can also be observed in the more viscous solvent glycerol (Fig. 4). The measured rotational speed for the depicted particle is 1.55 × 10−2 rad s−1. As for translational motion, the occurrence of a catalytically driven autonomous rotary motion (clockwise in the case illustrated, see ESI†) is attributed to the anisotropy of the structure with respect to cavitation points. The ability to decouple [O2] formation with directional control is an important feature of this approach as it enables directionality and catalyst activity to be addressed separately.
 | ||
| Fig. 4 Rotational movement of an 80 μm 1b-functionalised SiO2 particle in glycerol and its position at a) 0 s and b) 45 s. | ||
In conclusion, we describe the first molecular-based system in which catalytic conversion of chemical to mechanical energy induces autonomous movement of micro-sized particles. The exact mechanism by which propulsion of the particles takes place (recoil from O2 bubbles,13 interfacial tension gradient14) and the parameters governing the dynamics of the system need further investigation. The principle introduced here, to use a designed molecular catalyst to induce motion, is currently explored in the controlled motion of nanoparticles and functional molecules.
The authors thank the (WRB) Dutch Economy, Ecology, Technology (EET) program and the (JV) Departamento de Educación, Universidades e Investigación del Gobierno Vasco for financial support.
Notes and references
- Molecular Motors, ed. M. Schliwa, Wiley-VCH, Weinheim, 2002 Search PubMed.
- M. Venturi, A. Credi and V. Balzani, Molecular Devices and Machines – A Journey into the Nanoworld, Wiley-VCH, Weinheim, 2003 Search PubMed.
- (a) A. R. Pease, J. O. Jeppesen, J. F. Stoddart, Y. Luo, C. P. Collier and J. R. Heath, Acc. Chem. Res., 2001, 34, 433 CrossRef CAS; (b) R. Ballardini, V. Balzani, A. Credi, M. T. Gandolfi and M. Venturi, Acc. Chem. Res., 2001, 34, 445 CrossRef CAS; (c) C. M. Keaveney and D. A. Leigh, Angew. Chem. Int. Ed., 2004, 43, 1222 CrossRef CAS.
- (a) Z. Dominguez, T.-A. V. Khuong, H. Dang, C. N. Sanrame, J. E. Nunez and M. A. Garcia-Garibay, J. Am. Chem. Soc., 2003, 125, 8827 CrossRef; (b) Y. Kuwatani, G. Yamamoto and M. Iyoda, Org. Lett., 2003, 5, 3371 CrossRef CAS; (c) M. F. Hawthorne, J. I. Zink, J. M. Skelton, M. J. Bayer, C. Lui, E. Livshits, R. Baer and D. Neuhauser, Science, 2004, 303, 1849 CrossRef CAS.
- M. C. Jiménez, C. Dietrich-Buchecker and J.-P. Sauvage, Angew. Chem. Int. Ed., 2000, 39, 3284 CrossRef CAS.
- Molecular Switches, ed. B. L. Feringa, Wiley-VCH, Weinheim, 2001 Search PubMed.
- J. D. Badjić, V. Balzani, A. Credi, S. Silvi and J. F. Stoddart, Science, 2004, 303, 1845 CrossRef CAS.
- (a) N. Koumura, R. W. J. Zijlstra, R. A. van Delden, N. Harada and B. L. Feringa, Nature, 1999, 401, 15 CrossRef; (b) T. R. Kelly, H. De Silva and R. A. Silva, Nature, 1999, 401, 150 CrossRef CAS; (c) D. A. Leigh, J. K. Y. Wong, F. Dehez and F. Zerbetto, Nature, 2003, 424, 174 CrossRef CAS.
- P. Thordarson, E. J. A. Bijsterveld, A. E. Rowan and R. J. M. Nolte, Nature, 2003, 424, 915 CrossRef CAS.
- (a) J. E. Walker, Angew. Chem. Int. Ed., 1998, 37, 2308 CrossRef; (b) L. Stryer, Biochemistry, 4th edition, W. H. Freeman and Co., New York Search PubMed.
- B. L. Feringa, Acc. Chem. Res., 2001, 34, 504 CrossRef CAS.
- R.A. van Delden, N. Koumura, N. Harada and B.L. Feringa, Proc. Natl. Acad. Sci. USA, 2002, 99, 4945 CrossRef CAS.
- R. F. Ismagilov, A. Schwartz, N. Bowden and G. M. Whitesides, Angew. Chem. Int. Ed., 2002, 41, 652 CrossRef CAS.
- (a) W. F. Paxton, K. C. Kisler, C. O. Olmeda, A. Sen, S. K. St.Angelo, Y. Cao, T. E. Mallouk, P. E. Lammert and V. H. Crespi, J. Am. Chem. Soc., 2004, 126, 13424 CrossRef CAS; (b) J. M. Catchmark, S. Subramanian and A. Sen, Small, 2005, 1, 202 Search PubMed; (c) T. R. Kline, W. F. Paxton, T. E. Mallouk and A. Sen, Angew. Chem. Int. Ed., 2005, 44, 744 CrossRef CAS.
- S. Fournier-Bidoz, A. C. Arsenault, I. Manners and G. A. Ozin, Chem. Commun., 2005, 441 RSC.
- A. J. Wu, J. E. Penner-Hahn and V. L. Pecoraro, Chem. Rev., 2004, 104, 903 CrossRef CAS.
- Chemical formula: [C45H41Mn2N6O4][ClO4]; M: 939.18 g mol−1; Unit cell dimensions a = 15.226(2) Å (α = 90°), b = 12.924(2) Å (β = 90.157(9)°) , c = 10.944(2) Å (γ = 90°), V 2153.6(6) Å; T 130 K; monoclinic C2; Z = 2; μ (Mo Kα), cm−1 = 7.1; wR(F2) = 0.0778 for 4691 reflections with Fo2 ≥ 0 and R(F) = 0.0303 for 4481 reflections with Fo ≥ 4.0 σ(Fo) and 375 parameters, Flack parameter (X) = 0.428(15). CCDC number 268595. See http://www.rsc.org/suppdata/cc/b5/b505092h/ for crystallographic data in CIF or other electronic format.
- CH2Cl2, MeOH, CH3CN and glycerol were tested and gave satisfactory results.
- O2 evolution was measured in time using the gas burette method according to A. I. Unuchukwu and P. B. Mshelia, J. Chem. Ed., 1985, 62, 809 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: detailed experimental procedures for the synthesis of the ligand and complexes, spectroscopic and electrochemical data, observation of the moving particles and time videos representing the dynamic behaviour are shown as photographs (Fig. 3, 4) in this paper. See http://www.rsc.org/suppdata/cc/b5/b505092h/ |
| This journal is © The Royal Society of Chemistry 2005 |