Shell-crosslinked nanostructures from amphiphilic AB and ABA block copolymers of styrene-alt-(maleic anhydride) and styrene: polymerization, assembly and stabilization in one pot†
Simon
Harrisson
and
Karen L.
Wooley
*
Center for Materials Innovation and Department of Chemistry, Washington University, St Louis, MO 63130, USA. E-mail: klwooley@artsci.wustl.edu; Fax: +1 314 935 4481; Tel: +1 314 935 7136
First published on 12th May 2005
Abstract
Shell-crosslinked nanostructures having unusual rosette morphologies have been produced by a simple process from styrene and maleic anhydride.
Nanoparticles formed by the regioselective crosslinking of the outer regions of amphiphilic block copolymer micelles (by covalent coupling of chain segments constituting the shell1–5 or an intermediary layer6,7) have attracted great interest in recent years as well-defined nanoreactors,7 agents for encapsulation,3–5 transduction,3 and drug delivery,4 among other potential applications. The block copolymer precursors are programmed for multi-molecular assembly into complex nanostructures based upon their compositions and structures,8 which are controlled during synthesis using living polymerization techniques and via post-polymerization modifications. For physical and chemical compatibility purposes, protecting groups are often utilized during polymerization to establish the polymer structure and then are removed to reveal polar, reactive functionalities to promote supramolecular assembly and provide for covalent crosslinking.9
Armes and coworkers have developed an approach towards reducing the synthetic complexity of copolymer synthesis and micelle formation,2,6,7 whereby the direct production of micelles from block copolymers was accomplished by aqueous atom transfer radical polymerization of monomer units having pH- and thermally-tunable degrees of hydrophilicity. In another approach, the assembly of polymer mixtures into non-covalently crosslinked micelles has been followed by shell-crosslinking to establish robust nanomaterials by a block copolymer-free strategy.10 In this report, we present an alternative synthetic simplification strategy, which maintains the elegance of block copolymer assembly and utilizes a one-pot formation of well-defined, regioselectively-crosslinked nanostructures from commercially-available monomers.
To demonstrate this one-pot synthetic strategy, poly(maleic anhydride-alt-styrene)-block-styrene AB and ABA block copolymers were prepared from maleic anhydride (MA) and styrene (STY) via radical addition fragmentation chain transfer (RAFT) polymerization,11 hydrolyzed and organized into micellar assemblies upon the addition of water, and crosslinked by addition of a diamine in the presence of 1-[3-(dimethylamino)propyl]-3-ethylcarbodiimide hydrochloride (EDC), yielding shell-crosslinked nanostructures (Scheme 1). Each step within the overall one-pot methodology was performed independently to confirm the structure and composition of the products and to study its influence on the resulting assemblies and crosslinked nanostructures.
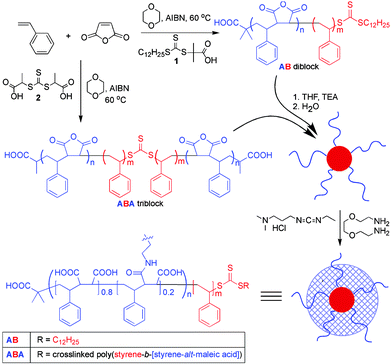 | ||
| Scheme 1 Synthesis of di- and tri-block poly(styrene-alt-maleic anhydride)-block-polystyrene copolymers and formation of micelles and shell-crosslinked nanoparticles. | ||
In the first stage, the block copolymers were synthesized from commodity monomers and without the need for sequential monomer additions and post-polymerization modification, allowing for significant cost and labor savings over current techniques for the synthesis of similar nanostructures. The propensity of MA and STY to form alternating copolymers regardless of their molar ratio is well-known. The living copolymerization of MA with STY has been reported under RAFT11,12 or nitroxide-mediated (NMRP)13,14 polymerization conditions. Of these, RAFT was selected, as it can be performed at temperatures lower than those required for NMRP,13 allowing the formation of MA–STY copolymers with structures much closer to the alternating ideal.11,12 The practical problems of synthesis, purification, and storage, commonly associated with RAFT agents, were ameliorated by the use of crystalline and virtually odorless RAFT agents, S-dodecyl S′-2-(2,2-dimethylacetic acid) trithiocarbonate, 1, and 2,2′-bis(propionic acid) trithiocarbonate, 2.15 Therefore, RAFT copolymerization of MA and STY was conducted in the presence of excess STY to afford an alternating copolymer initially, which was then extended by a homopolystyrene block segment, once the MA was exhausted. In this way, block copolymers were formed in which the length of the first STY-alt-MA block was determined by the ratio of MA to RAFT agent, while the length of the homo-STY block was determined by the ratio of excess STY to RAFT agent and the overall conversion.11,13,14
Di- and triblock copolymers with a range of compositions were conveniently prepared by dissolving appropriate amounts of maleic anhydride, styrene, RAFT agent 1 or 2, respectively, and 2,2′-azobisisobutyronitrile (AIBN) in 20 mL dioxane and heating for 21 h (reaching 75–96% conversion)
(Table 1). The resulting polymers exhibited two glass transition temperatures (Tg) at 77 and 170 °C, of varying intensities depending on the relative amounts of STY and MA, indicating the presence of microphase-separated domains in the bulk. Molecular weight distributions were unimodal and of narrow polydispersities, as observed by gel permeation chromatography (GPC). The architecture of the ABA triblock copolymer, 6, was confirmed by pyrolysis16 of the polymer (30 min at 250 °C), which resulted in cleavage at the central trithiocarbonate group to produce polymer chains having a number-average molecular weight half that of 6, and a narrow molecular weight distribution (Mn
= 11![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 100, PDI = 1.12).
100, PDI = 1.12).
| STY ∶ MA ∶ RAFT ∶ AIBN (initial ratios) | Conv.a | M n (theory)b | M n (GPC) | PDI | %STYc | Compositiond | |
|---|---|---|---|---|---|---|---|
| a Estimated by 1H NMR analysis of final reaction mixture. b Calculated from conv. × ([STY] + [MA])/[RAFT]. c From elemental analysis. d Ideal structure, calculated using %STY from elemental analysis, Mn from GPC, assuming no transitional region between poly(STY-alt-MA) and polySTY segments. e RAFT = 1. f RAFT = 2. | |||||||
| 3 | 250 ∶ 50 ∶ 1e ∶ 0.2 | 93% | 29200 |
23100 |
1.08 | 77.7 | (STY-alt-MA)52–STY118 |
| 4 | 200 ∶ 100 ∶ 1e ∶ 0.2 | 94% | 29100 |
29800 |
1.09 | 56.3 | (STY-alt-MA)117–STY25 |
| 5 | 300 ∶ 100 ∶ 1e ∶ 0.2 | 86% | 35700 |
35400 |
1.10 | 68.3 | (STY-alt-MA)109–STY113 |
| 6 | 150 ∶ 50 ∶ 1 f∶ 0.2 | 96% | 20200 |
20600 |
1.08 | 69.3 | (STY-alt-MA)28–STY63–(STY-alt-MA)28 |
In the cases of isolated block copolymers, aggregates were formed by slow addition (10 mL h−1) of an equal volume of water to a solution of copolymer in THF (2 mg mL−1) in the presence of a catalytic amount of triethylamine,‡ followed by dilution to 0.67 mg mL−1, dialysis against deionised water and, finally, dilution to 0.5 mg mL−1. The resulting solutions varied in appearance from colorless to bluish-white and were characterized by transmission electron microscopy (TEM), atomic force microscopy (AFM) and dynamic light scattering (DLS) (Table 2).† In solution, the assemblies of 3–5 were narrowly distributed in size, while the size distribution of 6 was significantly broader.
| Before crosslinking | After crosslinking | |||||||
|---|---|---|---|---|---|---|---|---|
| D h/nma | Polya | H/nmb | D/nmc | D h/nma | Polya | H/nm)b | D/nmc | |
| a Hydrodynamic volume (Dh) and polydispersity (Poly) measured by DLS. Numbers in parentheses represent standard error in the final digits. b Average height (and standard deviation) measured by AFM. c Average diameter (and standard deviation) measured by TEM. | ||||||||
| 3 | 41.2 (3) | 0.06 (2) | 50 (8) | 49 (7) | 84 (2) | 0.19 (5) | 48 (11) | 38 (8) |
| 4 | 45.0 (4) | 0.06 (2) | 39 (6) | 38 (7) | 100 (1) | 0.125 (2) | 49 (11) | 36 (8) |
| 5 | 58 (3) | 0.05 (4) | 56 (11) | 53 (11) | 102 (2) | 0.10 (2) | 62 (17) | 47 (11) |
| 6 | 30 (4) | 0.19 (1) | 23 (4) | 27 (6) | 44 (1) | 0.14 (2) | 32 (9) | 22 (6) |
When observed by TEM, assemblies of 3–5 frequently appeared to be composed of multiple subunits, arranged in a rosette pattern (Fig. 1A–C). This ordering was evident particularly for the structures formed from 5 (Fig. 1C) which were typically composed of 4, 5 or 6 subunits arranged circularly. The diameters by TEM (Table 2) were measured for the entire assemblies, rather than individual subunits. The agreement between diameters measured by DLS and by TEM suggests that these assemblies were present in solution and not an artefact of substrate adsorption. Higher-order assembly was not exhibited by 6, which formed much smaller aggregates without visible microstructure (Fig. 1D).
 | ||
| Fig. 1 TEM images of 3 (A), 4 (B), 5 (C) and 6 (D), showing rosette-like arrangements of subunits in assemblies of 5 (C). Scale bars are 100 nm. | ||
AFM of the micelles confirmed the dimensions observed by TEM and DLS. Higher-order structure was not observed by AFM, suggesting that the images obtained by TEM reveal the internal arrangement of polystyrene core domains in a continuous poly(styrene-alt-maleic acid) matrix. Intricate, internal segregation patterns have been observed for microscopic assemblies17 and are well known for block copolymers in the bulk, however, capture of such uniform structures within assemblies that are tens of nanometres in dimension is of significant interest.
Crosslinking was obtained via carbodiimide-mediated amidation,§ resulting in robust nanostructures. Dilution of the aqueous solutions with 9 volumes of THF caused disassembly of the noncrosslinked diblock copolymer assemblies, with the effect that no light scattering was observed from these solutions by DLS. In contrast, the crosslinked nanomaterials remained intact on dilution with THF. While the micelles of the triblock copolymer 6 did not dissociate completely on addition of THF, a clear difference was observed between crosslinked and noncrosslinked samples, with the crosslinked nanostructures showing greater resistance to THF-induced disassembly. Further confirmation of crosslinking was provided by IR analysis of the lyophilized crosslinked structures, which showed an amide carbonyl absorption band at 1636 cm−1. There was a significant increase in the hydrodynamic diameter and polydispersity in solution, as measured by DLS, suggesting increased hydrophilicity and swelling in aqueous solution upon incorporation of the hydrophilic crosslinker.
The structures formed from 3–5, as observed by TEM (Fig. 2), indicate that crosslinking was accompanied by an increase in the complexity of the microstructure (cf.Fig. 1). The mechanism of formation of these unusual structures is currently under investigation. The crosslinked morphologies from 6 were similar in appearance to their noncrosslinked precursors.
 | ||
| Fig. 2 Transmission electron micrographs of 3 (A), 4 (B), 5 (C), and 6 (D), crosslinked to a nominal crosslink density of 20%. Scale bars represent 100 nm. | ||
Finally, crosslinked nanoparticles were formed directly from the monomers without intervening purification steps.¶ The resulting mixture contained nanoparticles with Dh 164 ± 2 nm, and a polydispersity of 0.08 (measured by DLS). The persistence of the particles in 9 ∶ 1 THF ∶ H2O indicated that crosslinking had occurred.
The production of regioselectively crosslinked nanoparticles having well-defined, amphiphilic characteristics and rosette-like morphologies via a one-pot route from commodity monomers and involving intermediate block copolymer assemblies is a significant synthetic advance. Further studies are expected to lead to increased understanding of the structure of the internal sub-assemblies and the mechanism for their formation, ultimately, to allow for manipulation of those domains and enhanced complexity.
This material is based upon work supported by the National Science Foundation under Grant No. 0301833. Mass spectrometry was provided by the Washington University Mass Spectrometry Resource, an NIH Research Resource (Grant No. P41RR0954). The authors thank Mr G. Michael Veith (Washington University Electron Microscopy Laboratory) for assistance with TEM.
Notes and references
- V. Bütün, N. C. Billingham and S. P. Armes, J. Am. Chem. Soc., 1998, 120, 12135 CrossRef.
- Y. Ma, Y. Tang, N. C. Billingham, S. P. Armes, A. L. Lewis, A. W. Lloyd and J. P. Salvage, Macromolecules, 2003, 36, 3475 CrossRef CAS.
- K. L. Wooley, J. Polym. Sci., Part A: Polym. Chem., 2000, 38, 1397 CrossRef CAS; H. M. Kao, R. D. O'Connor, A. K. Mehta, H. Huang, B. Poliks, K. L. Wooley and J. Schaefer, Macromolecules, 2001, 34, 544 CrossRef CAS; H. Huang, K. L. Wooley and J. Schaefer, Macromolecules, 2001, 34, 547 CrossRef CAS.
- M. L. Becker, E. E. Remsen, D. Pan and K. L. Wooley, Bioconjugate Chem., 2004, 15, 699 CrossRef CAS; M. L. Becker, L. O. Bailey and K. L. Wooley, Bioconjugate Chem., 2004, 15, 710 CrossRef CAS.
- M. L. Becker, J. Liu and K. L. Wooley, Biomacromolecules, 2005, 6, 220 CrossRef CAS; D. Pan, J. L. Turner and K. L. Wooley, Macromolecules, 2004, 37, 7109 CrossRef CAS.
- V. Bütün, X.-S. Wang, M. V. d. P. Báñez, K. L. Robinson, N. C. Billingham, S. P. Armes and Z. Tuzar, Macromolecules, 2000, 33, 1 CrossRef; S. Liu and S. P. Armes, J. Am. Chem. Soc., 2001, 123, 9910 CrossRef CAS.
- S. Liu, J. V. M. Weaver, M. Save and S. P. Armes, Langmuir, 2002, 18, 8350 CrossRef CAS.
- O. Terreau, L. Luo and A. Eisenberg, Langmuir, 2003, 19, 5601 CrossRef CAS; S. E. Burke and A. Eisenberg, Langmuir, 2001, 17, 6705 CrossRef CAS; D. J. Pochan, Z. Chen, H. Cui, K. Hales, K. Qi and K. L. Wooley, Science, 2004, 306, 94 CrossRef CAS; Z. Li, E. Kesselman, Y. Talmon, M. A. Hillmyer and T. P. Lodge, Science, 2004, 306, 98 CrossRef CAS.
- H. Huang, T. Kowalewski, E. E. Remsen, R. Gertzmann and K. L. Wooley, J. Am. Chem. Soc., 1997, 119, 11653 CrossRef CAS.
- M. Kuang, H. Duan, J. Wang, D. Chen and M. Jiang, Chem. Commun., 2003, 496 RSC; X. Liu, M. Jiang, S. Yang, M. Chen, D. Chen, C. Yang and K. Wu, Angew. Chem., Int. Ed., 2002, 41, 2950 CrossRef CAS; M. Wang, M. Jiang, F. Ning, D. Chen, S. Liu and H. Duan, Macromolecules, 2002, 35, 5980 CrossRef CAS; X. F. Yuan, M. Jiang, H. Y. Zhao, M. Wang, Y. Zhao and C. Wu, Langmuir, 2001, 17, 6122 CrossRef CAS; Y. Zhang, M. Jiang, J. Zhao, J. Zhou and D. Chen, Macromolecules, 2004, 37, 1537 CrossRef CAS.
- M.-Q. Zhu, L.-H. Wei, M. Li, L. Jiang, F.-S. Du, Z.-C. Li and F.-M. Li, Chem. Commun., 2001, 365 RSC.
- H. de Brouwer, M. A. J. Schellekens, B. Klumperman, M. J. Monteiro and A. L. German, J. Polym. Sci., Part A: Polym. Chem., 2000, 38, 3596 CrossRef CAS; X. Hao, M. H. Stenzel, C. Barner-Kowollik, T. P. Davis and E. Evans, Polymer, 2004, 45, 7401 CrossRef CAS; E. Chernikova, P. Terpugova, C. Bui and B. Charleux, Polymer, 2003, 44, 4101 CrossRef CAS; F. S. Du, M.-Q. Zhu, H. Q. Guo, Z.-C. Li, F.-M. Li, M. Kamachi and A. Kajiwara, Macromolecules, 2002, 35, 6739 CrossRef CAS.
- D. Benoit, C. J. Hawker, E. E. Huang, Z. Lin and T. P. Russell, Macromolecules, 2000, 33, 1505 CrossRef CAS.
- E. S. Park, M.-N. Kim, I.-M. Lee, H.-S. Lee and J.-S. Yoon, J. Polym. Sci., Part A: Polym. Chem., 2000, 38, 2239 CrossRef CAS.
- J. T. Lai, D. Filla and R. Shea, Macromolecules, 2002, 35, 6754 CrossRef CAS.
- A. Postma, T. P. Davis, G. Moad and M. S. O'Shea, Macromolecules, 2005, 38, in press Search PubMed.
- Z. Lu, G. Liu and F. Liu, Macromolecules, 2001, 34, 8814 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Full experimental details, AFM images and DLS correlation functions. See http://www.rsc.org/suppdata/cc/b5/b504313a/ |
| ‡ In the absence of triethylamine, precipitation occurred on water addition. |
| § In a typical procedure, 1,2-ethylenedioxy bis(ethylamine) (10 mol% relative to maleic anhydride units) was added to a micellar solution of polymer. After stirring for 20 min, EDC was added (20 mol% relative to maleic anhydride units). The solution was stirred overnight at RT, then dialyzed for 3 days against deionised H2O to remove the urea byproduct. |
| ¶ MA (0.5 g) and STY (1.5 g) were heated at 60 °C in the presence of 2.0 g dioxane, 18 mg 1, and a trace of AIBN for a period of 16 h under N2. The resulting mixture (conversion = 89% by NMR) was dissolved in 1 L of THF. 1,2-Ethylenedioxy bis(2-ethylamine) (18.9 mg, 10 mol% relative to maleic anhydride) was added with vigorous stirring to a 250 mL aliquot of this solution, followed immediately by 250 mL nanopure water. The THF was removed by evaporation in vacuo (at RT) and the resulting micellar solution was diluted to 1 L with nanopure water. |
| This journal is © The Royal Society of Chemistry 2005 |