A novel one-pot three-component synthesis of 3-halofurans and sequential Suzuki coupling†
Alexei S.
Karpov
,
Eugen
Merkul
,
Thomas
Oeser
and
Thomas J. J.
Müller
*
Organisch-Chemisches Institut der Ruprecht-Karls-Universität Heidelberg, Im Neuenheimer Feld 270, D-69120 Heidelberg, Germany. E-mail: Thomas_J.J.Mueller@urz.uni-heidelberg.de; Fax: +49(0)6221546579; Tel: +49(0)6221546207
First published on 6th April 2005
Abstract
A novel sequence of Sonogashira coupling and electrophilic addition to an ynone, with concomitant deprotection and cyclocondensation, opens a new one-pot synthesis of 3-halofurans; the method can be readily elaborated to a new sequential Sonogashira–addition–cyclocondensation–Suzuki reaction to furnish 2,3,5-trisubstituted furans in a one-pot fashion.
Furans are ubiquitous structural units in numerous natural products,1 in pharmaceuticals,2 and even in photonic chromophores.3 Among various syntheses of furans,4 the two major approaches5 are either based upon the construction of the furan ring starting from acyclic precursors6 or substitution reactions on the furan core. In particular, by regiospecific substitutions, halofurans are ideal starting materials, either as electrophiles in cross-coupling reactions7 or, via halogen–metal exchange, as nucleophiles for subsequent electrophilic trapping.8 However, efficient and concise syntheses of 3-substituted halofurans are still a methodological challenge.9,10 As part of our program directed to develop new one-pot multi-component heterocycle syntheses initiated by transition metal catalyzed alkyne coupling,11 here, we communicate a novel one-pot three-component synthesis of 3-halofurans and sequential cross-coupling, still in a one-pot fashion.
Recently, we have developed a modification of the Sonogashira coupling of acid chlorides and terminal alkynes to give alkynones,11 where only one stoichiometric equivalent of triethylamine, necessary as hydrochloric acid scavenging base, is applied. Therefore, the reaction medium becomes essentially base free, now setting the stage for acid catalyzed consecutive steps. Ynones possess an enormous potential as key intermediates in heterocycle synthesis.12 Hence, we reacted benzoyl chloride (1a) and the tetrahydropyranyl propargyl ether (2a) under modified Sonogashira conditions, followed by the addition of NaCl and p-tolylsulfonic acid (PTSA) in methanol, to give, through the intermediacy of an γ-hydroxy alkynone,13 4-chloro-2-phenylfuran (3a) in 63% yield (Scheme 1).
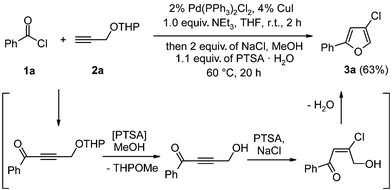 | ||
| Scheme 1 Coupling–addition–cyclocondensation sequence to 4-chlorofuran 3a. | ||
This novel sequence can be rationalized as a cross-coupling furnishing a THP-protected 3-hydroxy alkynone that is solvolyzed under acid catalysis to give rise to the γ-hydroxy alkynone. Acid-assisted Michael addition of HCl and subsequent cyclocondensation conclude the three-component sequence to give the 4-chlorofuran 3a.
According to these optimal conditions, with the extension to using sodium iodide as a halide source, various acid chlorides 1 and tetrahydropyranyl propargyl ethers 2 can be successfully transformed into 3-halofurans 3 in a one-pot coupling–addition–cyclocondensation sequence (Scheme 2, Table 1).14
 | ||
| Scheme 2 Coupling–addition–cyclocondensation synthesis of 3-halofurans 3. | ||
| Entry | Acid chloride 1 | Alkyne 2 | 3-Halofuran 3 (yield) |
|---|---|---|---|
| a 2.0 equiv. of NaCl, 60 °C, 20 h. b 5 equiv. of NaI, r.t., 2 h. | |||
| 1a | R1 = Ph (1a) | R2 = H (2a) | 3a (R1 = Ph , R2 = H, Hal = Cl, 63%) |
| 2a | R1 = p-MeOC6H4 (1b) | 2a | 3b (R1 = p-MeOC6H4, R2 = H, Hal = Cl, 71%) |
| 3a | 1a | R2 = Et (2b) | 3c (R1 = Ph, R2 = Et, Hal = Cl, 70%) |
| 4a | R1 = 2-thienyl (1c) | 2b | 3d (R1 = 2-thienyl, R2 = Et, Hal = Cl, 59%) |
| 5a | R1
= PhCH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH (1d) CH (1d) |
2b |
3e
(R1
= PhCHCH, R2
= Et, Hal = Cl, 73%) |
| 6a | R1 = 1-cyclohexenyl (1e) | 2a | 3f (R1 = 1-cyclohexenyl, R2 = H, Hal = Cl, 64%) |
| 7b | 1a | 2a | 3g (R1 = Ph, R2 = H, Hal = I, 63%) |
| 8b | 1b | 2a | 3h (R1 = p-MeOC6H4, R2 = H, Hal = I, 63%) |
| 9b | R1 = p-NO2C6H4 (1f) | 2a | 3i (R1 = p-NO2C6H4, R2 = H, Hal = I, 40%) |
| 10b | 1a | 2b | 3j (R1 = Ph, R2 = Et, Hal = I, 72%) |
The structure of the 3-halofurans 3 is unambiguously supported by an X-ray structure analysis for 3i (Fig. 1).‡
 | ||
| Fig. 1 Molecular structure of 3i (R1 = p-NO2C6H4, R2 = H, Hal = I). Only 1 of 4 independent molecules is shown. The enumeration is adjusted. | ||
Methodologically, this new one-pot three-component synthesis of 3-halofurans proceeds efficiently under mild conditions with a wide variety of electronically diverse acid chlorides. Applying NaI as a halide source leads to even milder reaction conditions and shorter reaction times, now giving extremely valuable 3-iodofurans. Therefore, due to the acid sensitivity of iodofurans, this methodology has significant advantages over existing protocols using HI as an acid.
Finally, as a showcase for the highly topical field of sequential catalysis15 we probed a sequential Sonogashira–addition–cyclocondensation–Suzuki reaction where the same catalyst system should be applied for two consecutive, significantly different, cross-coupling reactions in the same reaction vessel. Therefore, upon consecutive reactions of acid chlorides 1 and tetrahydropyranyl propargyl ethers 2, NaI and PTSA, and addition of 1.05 equiv. of boronic acids 4 and sodium carbonate, the substituted 3-arylfurans 5 were obtained in decent yields (Scheme 3).16
 | ||
| Scheme 3 Sequential Sonogashira–addition–cyclocondensation–Suzuki synthesis of substituted 3-arylfurans 5. | ||
The new one-pot Sonogashira–addition–cyclocondensation–Suzuki synthesis of substituted 3-arylfurans 5 proceeds in reasonable yields that are almost comparable (one-pot sequence to 5a: 50%) with a stepwise procedure (overall yield of 5a: 45%).
In conclusion, we have developed a novel consecutive three-component coupling–addition–cyclocondensation synthesis of 3-halofurans, highly versatile building blocks in organic synthesis. In addition, a new sequential Sonogashira–addition–cyclocondensation–Suzuki multi-component furan synthesis was readily elaborated as a new diversity-oriented consecutive multi-component access to substituted 3-arylfurans. Studies addressing the scope of this sequence to enhance molecular diversity are currently under investigation.
The authors gratefully acknowledge DFG (Graduiertenkolleg 850), MORPHOCHEM AG, Fonds der Chemischen Industrie, and Dr Otto-Röhm Gedächtnisstiftung, and cordially thank Ms Michaela Schmitt for experimental assistance.
Notes and references
-
B. A. Keay and P. W. Dibble, in Comprehensive Heterocyclic Chemistry II, ed. A. R. Katritzky, C. W. Rees and E. F. V. Scriven, Elsevier, Oxford, 1997, vol. 2, pp. 395 Search PubMed
.
- For pharmaceuticals, see e.g.: D. S. Mortensen, A. L. Rodrigues, K. E. Karlson, J. Sun, B. S. Katzenellenbogen and J. A. Katzenellenbogen, J. Med. Chem., 2001, 44, 3838 Search PubMed
- C. Liu and T. Luh, Org. Lett., 2002, 4, 4305 CrossRef CAS
- For reviews, see e.g.: R. C. D. Brown, Angew. Chem. Int. Ed., 2005, 44, 850 Search PubMed
- For recent reviews, see e.g.: T. L. Gilchrist, J. Chem. Soc., Perkin Trans. 1, 1999, 2849 Search PubMed
- For Pd-catalyzed transformations of acyclic precursors to furans, see e.g.: A. Jeevanandam, A. Ghule and Y.-C. Ling, Curr. Org. Chem., 2002, 6, 841 Search PubMed
- For investigations on cross-coupling of halofurans, see e.g.: M. W. Hooper and J. F. Hartwig, Organometallics, 2003, 22, 3394 Search PubMed
- For the preparation of 3-lithiofuran, see e.g.: C. C. Bond and M. Hooper, Synthesis, 1974, 443 Search PubMed
- For the recent synthesis of 3-substituted 4-chlorofurans, see e.g.: R. N. Ram and I. Charles, Chem. Commun., 2003, 2267 Search PubMed
- For the recent synthesis and further transformations of β-iodofurans, see e.g.: G. M. M. El-Taeb, A. B. Evans, S. Jones and D. W. Knight, Tetrahedron Lett., 2001, 42, 5945 Search PubMed
- A. S. Karpov, T. Oeser and T. J. J. Müller, Chem. Commun., 2004, 1502 RSC
- For heterocycle synthesis from ynones, see e.g.: M. C. Bagley, D. D. Hughes and P. H. Taylor, Synlett, 2003, 259 Search PubMed
- The successful conversion of γ-hydroxy alkynones into halofurans has been reported by: D. Obrecht, Helv. Chim. Acta, 1989, 72, 447 Search PubMed
-
Typical procedure (compound 3j): 14 mg (0.02 mmol) of Pd(PPh3)2Cl2 and 7 mg (0.04 mmol) of CuI were dissolved in 5 mL of degassed THF. Then 141 mg (1.0 mmol) of 1a, 168 mg (1.0 mmol) of 2b, and 0.14 mL (1.00 mmol) of triethylamine were successively added to the solution and the mixture was stirred for 2 h at room temperature. Then 750 mg (5.00 mmol) of sodium iodide, 209 mg (1.10 mmol) of p-tolylsulfonic acid monohydrate and 3 mL of methanol were added and stirring at room temperature was continued for 2 h. After aqueous work up with a saturated solution of NaHCO3 and Na2SO3 and chromatography on neutral aluminium oxide (hexane–ethyl acetate 9∶1–20∶1), 215 mg (72%) of pure 3j were obtained as a light yellow oil. 1H NMR (acetone-d6, 300 MHz): δ 1.26 (t, J
= 7.7 Hz, 3 H), 2.76 (q, J
= 7.7 Hz, 2 H), 6.89 (s, 1 H), 7.26–7.32 (m, 1 H), 7.38–7.45 (m, 2 H), 7.66–7.71 (m, 2 H). 13C NMR (acetone-d6, 75 MHz): δ 12.8 (CH3), 21.6 (CH2), 63.9 (Cquat), 113.6 (CH), 124.2 (CH), 128.5 (CH), 129.7 (CH), 131.0 (Cquat), 154.4 (Cquat), 158.1 (Cquat). Calc. HRMS for C12H11IO: 297.9855. Found: 297.9861
- For excellent recent reviews, see e.g.: A. Ajamian and J. L. Gleason, Angew. Chem. Int. Ed., 2004, 43, 3754 Search PubMed
-
Typical procedure (compound 5a): 35 mg (0.05 mmol) of Pd(PPh3)2Cl2 and 7 mg (0.04 mmol) of CuI were dissolved in 5 mL of degassed THF. Then 171 mg (1.00 mmol) of 1b, 141 mg (1.00 mmol) of 2a, and 0.14 mL (1.00 mmol) of triethylamine were successively added to the solution and the mixture was stirred for 2 h at room temperature. Then 750 mg (5.00 mmol) of sodium iodide, 209 mg (1.10 mmol) of p-tolylsulfonic acid monohydrate and 3 mL of methanol were added and stirring at room temperature was continued for 2 h. Then 4 mL (8 mmol) of a 2 M solution of aqueous sodium carbonate and 128 mg (1.05 mmol) of boronic acid 4a were added and the mixture was heated at 90 °C for 28 h. After aqueous work up and chromatography on silica gel, 115 mg (50%) of the analytically pure 2-substituted 3-phenylfuran 5a were obtained as a colorless solid. Rf
= 0.42 (hexane–ethyl acetate 9∶1). Mp 129 °C. 1H NMR (acetone-d6, 300 MHz): δ 3.84 (s, 3 H), 7.02 (d, J
= 8.8 Hz, 2 H), 7.16 (d, J
= 0.7 Hz, 1 H), 7.24–7.31 (m, 1 H), 7.36–7.44 (m, 2 H), 7.63–7.68 (m, 2 H), 7.72 (d, J
= 8.8 Hz, 2 H), 8.02 (d, J
= 0.7 Hz, 1H). 13C NMR (acetone-d6, 75 MHz): δ 55.6 (CH3), 103.3 (CH), 115.1 (CH), 124.5 (Cquat), 126.1 (CH), 126.5 (CH), 127.8 (CH), 129.3 (Cquat), 129.6 (CH), 133.4 (Cquat), 138.6 (CH), 155.8 (Cquat), 160.4 (Cquat). EI MS [m/z
(%)]: 250 (M+, 100), 235 (M+
− CH3, 14), 221 (M+
− CHO, 15). Anal calc. for C17H14O2
(250.30): C 81.58, H 5.64. Found: C 81.20, H 5.63
Footnotes |
| † Electronic supplementary information (ESI) available: experimental procedures and characterization for compounds 3 and 5. See http://www.rsc.org/suppdata/cc/b5/b502324f/ |
‡ Crystal data for 3i: C10H6INO3, M
= 315.1, triclinic, space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) , a
= 8.2679(1), b
= 11.0675(1), c
= 22.2477(2)
Å, α
= 84.927(1)°, β
= 83.749(1)°, γ
= 88.385(1)°, V
= 2015.39(4)
Å3, T
= 200(2) K, Z
= 8, ρ
= 2.077 g cm−3, crystal dimensions 0.50 × 0.34 × 0.30 mm3, Mo Kα radiation, μ
= 3.162 mm−1, λ
= 0.71073 Å. There are four independent molecules in the asymmetric unit. Data were collected on a Bruker Smart APEX diffractometer and a total of 9160 of the 20833 reflections were unique [R(int)
= 0.0201]. Refinement on F2, wR2
= 0.049 (observed reflections), R1
= 0.021 for [I > 2σ(I)]. CCDC 260725. See http://www.rsc.org/suppdata/cc/b5/b502324f/ for crystallographic data in CIF or other electronic format. , a
= 8.2679(1), b
= 11.0675(1), c
= 22.2477(2)
Å, α
= 84.927(1)°, β
= 83.749(1)°, γ
= 88.385(1)°, V
= 2015.39(4)
Å3, T
= 200(2) K, Z
= 8, ρ
= 2.077 g cm−3, crystal dimensions 0.50 × 0.34 × 0.30 mm3, Mo Kα radiation, μ
= 3.162 mm−1, λ
= 0.71073 Å. There are four independent molecules in the asymmetric unit. Data were collected on a Bruker Smart APEX diffractometer and a total of 9160 of the 20833 reflections were unique [R(int)
= 0.0201]. Refinement on F2, wR2
= 0.049 (observed reflections), R1
= 0.021 for [I > 2σ(I)]. CCDC 260725. See http://www.rsc.org/suppdata/cc/b5/b502324f/ for crystallographic data in CIF or other electronic format. |
| This journal is © The Royal Society of Chemistry 2005 |