Poly(ionic liquid)s: a new material with enhanced and fast CO2 absorption†
Jianbin
Tang‡
ab,
Huadong
Tang
a,
Weilin
Sun
b,
Henry
Plancher
a,
Maciej
Radosz
a and
Youqing
Shen
*a
aDepartment of Chemical & Petroleum Engineering, University of Wyoming, Laramie, WY 82071, USA. E-mail: sheny@uwyo.edu
bDepartment of Polymer Science & Engineering, Zhejiang University, Hanzhou, China
First published on 27th May 2005
Abstract
Novel sorbent and membrane materials for CO2 separation, poly(ionic liquid)s made from ionic liquid monomers, poly[p-vinylbenzyltrimethyl ammonium tetrafluoroborate] (P[VBTMA] [BF4]) and poly[2-(methacryloyloxy)ethyltrimethylamnonium tetrafluoroborate] (P[MATMA][BF4]) have absorption capacities 7.6 and 6.0 times of those of room-temperature ionic liquids, e.g. [bmim][BF4], respectively, with reversible and fast sorption and desorption.
Ionic liquids are organic salts that are liquids at low temperatures.1 Recently, CO2 has been shown to be remarkably soluble in imidazolium-based ionic liquids.2–11 For instance, at 15 bar of CO2 pressure, the CO2 solubility in 1-butyl-3-methylimidazolium hexafluorophosphate ([bmim][PF6]) is about 23 mol%.3 The CO2 solubility can be tuned by choice of cations, anions, and substituents of the ionic liquids, and anions play a major role.8 For example, using fluorine-containing anions (e.g. bis(trifluoromethylsulfonyl)imide, Tf2N)8 or cations,11 and introducing amine groups12 could increase the CO2 solubility. Ionic liquids are also impregnated on porous supports to develop supported liquid membranes (SLM). These membranes have high CO2 selectivity and permeance.13 Therefore, ionic liquids may be very useful in CO2 separation for carbon sequestration.
Recently, we found that simply making the ionic liquids into polymeric forms significantly increased the CO2 sorption capacity compared with ionic liquids. Especially, the polymers of tetraalkylammonium-based ionic liquids have CO2 sorption capacities 6.0–7.6 times of those of room temperature ionic liquids. The CO2 sorption and desorption of the polymer solids are very fast, and the desorption is completely reversible. These polymers are very prospective as sorbent and membrane materials for CO2 separation.
The ionic liquid monomers were prepared by anion exchange reactions and their corresponding polymers were synthesized by free radical polymerization. The structures of the polymers, poly[p-vinylbenzyltrimethylammonium tetrafluoroborate] (P[VBTMA] [BF4]), poly[2-(methacryloyloxy)ethyltrimethylammonium tetrafluoroborate] (P[MATMA][BF4]), poly[1-(p-vinylbenzyl)-3-butylimidazolium tetrafluoroborate](P[VBBI][BF4]) or bis(trifluoromethylsulfonyl)imide (P[VBBI][Tf2N]), and poly[1-(2-methacryloyloxy)ethyl-3-butylimidazolium tetrafluoroborate] (P[MABI][BF4]) are shown in Scheme 1. The X-ray diffraction and DSC data show that all the polymers are amorphous. They have glass transition temperatures at 215 °C (P[VBTMA][BF4]), 166 °C (P[MATMA][BF4]), 86 °C (P[VBBI][BF4]), 30 °C (P[VBBI] [Tf2N]) and 69 °C (P[MABI][BF4]), respectively. At room temperature, all polymers are easily crushed into fine powders. The specific surface areas of the particles measured by BET are 0.46 m2 g−1 for P[VBTMA][BF4], 20.5 m2 g−1 for P[MATMA][BF4], 0.29 m2 g−1 for P[VBBI][BF4], 0.77 m2 g−1 for P[MABI][BF4]. SEM shows that the particles of P[MATMA][BF4] are porous, and the others are nonporous.
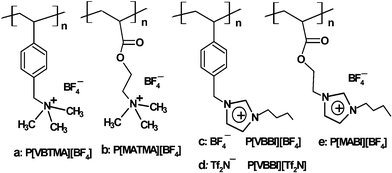 | ||
| Scheme 1 The structures of the poly(ionic liquid)s. | ||
The gas sorption of the polymers was measured under local ambient pressure (592.3 mmHg, Laramie, Wyoming). A detailed description of the apparatus and the experimental procedure are given in the Supporting Information†. The buoyancy effects were corrected according to the reported method.14 The gas was dried by passing through two columns of phosphorus peroxide (P2O5). A layer of P2O5 was also put on the bottoms of the two chambers of the microbalance. The system was validated by measuring the CO2 absorption of an ionic liquid, 1-n-butyl-3-methyl imidazolium tetrafluoroborate ([bmim][BF4]). The measured CO2 absorption capacity was 0.261 wt% (1.34 mol%) at 592.3 mmHg CO2 and 22 °C, consistent with the reported.8
Fig. 1. shows the CO2 sorption kinetics of the polymers, their corresponding monomers and a room-temperature ionic liquid [bmim][BF4]. At equilibrium, the polymers took up 10.22 mol% (P[VBTMA][BF4]), 7.99 mol% (P[MATMA][BF4]), 2.27 mol% (P[VBBI][BF4] or P[VBBI][Tf2N]), and 1.80 mol% (P[MABI][BF4]) of CO2 in terms of their monomer units. In comparison, room temperature ionic liquid [bmim][BF4] only absorbed 1.34 mol% of CO2 under the same conditions. Their monomers, [VBTMA][BF4] (f), [MATMA][BF4] (g), and [VBBI][BF4] (h) did not absorb CO2 at all because of their crystalline structures. The [MABI][BF4] monomer is a liquid at room temperature having the same CO2 absorption capacity as [bmim][BF4] (Fig. 1). This comparison shows that simply making the ionic liquids into polymeric forms can significantly increase the CO2 sorption capacity. These CO2 sorption capacities are also significantly higher than other polymer solids such as polymethacrylates, polystyrene and polycarbonates.15–17
![CO2 sorption of the polymers (a–e) in Scheme 1, their corresponding monomers [VBTMA][BF4](f), [MATMA][BF4](g), [VBBI][BF4](h), ([MABI][BF4](i), and ionic liquid [bmim][BF4]
(j) as a function of time (592.3 mmHg CO2, 22 °C).](/image/article/2005/CC/b501940k/b501940k-f1.gif) | ||
| Fig. 1 CO2 sorption of the polymers (a–e) in Scheme 1, their corresponding monomers [VBTMA][BF4](f), [MATMA][BF4](g), [VBBI][BF4](h), ([MABI][BF4](i), and ionic liquid [bmim][BF4] (j) as a function of time (592.3 mmHg CO2, 22 °C). | ||
The anion, cation and polymer backbone affect the CO2 sorption capacity, but the type of cation plays a major role. For example, tetraalkylammonium-based poly(ionic liquid)s (a,b) had much higher CO2 sorption capacity than the imidazolium-based poly(ionic liquid)s (c,d,e). This may be due to the tetraalkylammonium cation having a higher positive charge density and thus stronger interaction with CO2. In contrast, the positive charge of the imidazolium is delocalized. The anions have little effects on the CO2 sorption capacity. For example, P[VBBI][BF4] (c) and P[VBBI][Tf2N] (d) had the same CO2 sorption capacity. This is in contrast to the findings in the room temperature ionic liquids, in which anion is the main parameter affecting the CO2 solubility in ionic liquids, and Tf2N− anions enhance the CO2 solubility.8 With the same cations and anions, the polymers with polystyrene backbone had higher CO2 sorption capacity than those with polymethylmethacrylate backbones.
The sorption rates of these polymer particles are very fast (Fig. 1). It took only several minutes for the polymers to reach their 95% sorption capacities. In contrast, it took more than 400 min for room temperature ionic liquids [MABI][BF4] and [bmim][BF4] to reach their equilibrium. The fast sorption rates of the polymers is not solely because of their small particle sizes (∼100 µm). Fig. 1 shows that the CO2 sorption kinetics was not affected by their surface areas (i.e. particle sizes). The particles with specific areas less than 1 m2 g−1 took up CO2 as fast as porous P[MATMA][BF4] particles having specific areas of 20 m2 g−1. Additionally, the larger particles (∼500 µm) of P[VBBI][Tf2N] still took up CO2 very fast (Fig. 1d). In contrast, when BF4− anions in P[VBBI][BF4] were replaced with Cl− anions, the resulting poly[1-(4-vinylbenzyl)-3-butylimidazolium chloride] sorbed CO2 very slowly (Supporting Information†) even with the same particle sizes. Therefore, the fast CO2 sorption is characteristic of the poly(ionic liquid)s.
The desorption of the poly(ionic liquid)s is also fast (Fig. 2A). Under vacuum, the polymers released CO2 in less than 15 min. The desorption was complete, and no change in sorption/desorption kinetics and sorption capacity was observed after four cycles of sorption/desorption experiments, suggesting that the sorption/desorption was completely reversible. In contrast, the desorption of ionic liquids (e.g.[bmim][BF4]) was also very slow (Fig. 2B).
![(A) Cycles of CO2 sorption (592.3 mmHg CO2, 22 °C) and desorption under vacuum of (a) P[VBTMA][BF4] and (b) P[MATMA][BF4], and (B) CO2 absorption (592.3 mmHg CO2, 22 °C) and desorption under vacuum of [bmim][BF4].](/image/article/2005/CC/b501940k/b501940k-f2.gif) | ||
| Fig. 2 (A) Cycles of CO2 sorption (592.3 mmHg CO2, 22 °C) and desorption under vacuum of (a) P[VBTMA][BF4] and (b) P[MATMA][BF4], and (B) CO2 absorption (592.3 mmHg CO2, 22 °C) and desorption under vacuum of [bmim][BF4]. | ||
The CO2 sorption of P[VBTMA][BF4] as a function of pressure is shown in Fig. 3. The sorption capacity increased with the increase of the CO2 pressure. For example, P[VBTMA][BF4] absorbed 44.8 mol% of CO2 (in terms of its monomer units) at 12 atm. of CO2 pressure, much higher than that of room temperature ionic liquids.8
![CO2 sorption capacities of P[VBTMA][BF4] at different CO2 pressures (22 °C).](/image/article/2005/CC/b501940k/b501940k-f3.gif) | ||
| Fig. 3 CO2 sorption capacities of P[VBTMA][BF4] at different CO2 pressures (22 °C). | ||
The enhanced sorption capacity and fast sorption/desorption rates of the poly(ionic liquid)s were unexpected because the polymers are solid at room temperature. The calculated CO2-adsorption of P[VBTMA][BF4] assuming a monolayer of CO2 on the polymer particle surface was about 0.02 wt%, much less than the measured CO2 sorption capacities (1.70 wt%) at 592.3 mmHg of CO2. This indicates that the bulk of the polymer particles plays a major role in the CO2 sorption. Therefore, the CO2 sorption of the polymer particles involves more absorption (the bulk) but less adsorption (the surface). The fast absorption/desorption rates indicate that the diffusion of CO2 in the polymer solids is very fast. At present, the underlying mechanism of the enhanced absorption capacity and fast sorption/desorption of the polymer solids are still under investigation.
The CO2 absorption of the polymers is very selective. There was no weight gain when the polymers were exposed to N2 or O2 under the same conditions (Supporting Information†). Moisture could slightly decease the CO2 absorption capacity. For example, wet P[VBTMA][BF4] with 13.8 mol% water had a CO2 absorption capacity of 7.9 mol%, lower than that of dry P[VBTMA][BF4].
In summary, we demonstrate that the poly(ionic liquid)s are a novel polymer materials that selectively absorb CO2 with higher absorption capacity and faster absorption/desorption rates than room-temperature ionic liquids. These characters make these polymers exceptionally promising as absorbent and membrane materials for CO2 separation. The poly(ionic liquid) membranes for CO2 separation will be reported soon.
The ionic liquid monomers were prepared by anion exchange reaction of the corresponding chloride salts with NaBF4, NaPF6 or lithium trifluoromethane sulfonamide(LiTf2N).† An example of the synthesis of [MATMA][BF4] is as follows: Aqueous 2-(methylacryloyloxy)ethyltrimethylammonium chloride solution (75 wt%) (30 ml, 0.12 mol) was added into a 250 ml flask. After the water was removed under vacuum, NaBF4 (14.5 g, 0.132 mol) and CH3CN (150 ml) were added to the flask. The mixture was stirred over night. The salt dissolved gradually and a white precipitate formed. The precipitate was removed by filtration. The filtrate was concentrated, and then poured into ether. The white crystals were collected and dried under vacuum at room temperature (28 g, 90%). Similarly, [VBTMA][BF4] was synthesized from p-vinylbenzyltrimethylammonium chloride in a yield of 93%.
The polymers were synthesized via free radical polymerization. A general procedure is as follows: Ionic liquid monomers (2 g), AIBN (20 mg) and DMF (4 ml) were charged into a reaction tube. The tube was tightly sealed and degassed. The tube was immersed in an oil bath at 60 °C for 6 h. After polymerization, the solution of polymer was poured in methanol to precipitate the polymer. The polymer was dried under vacuum at 60 °C. The yield was 75% for P[VBBI][BF4], 68% for P[VBBI][Tf2N], 70% for P[MABI][BF4], 90% for P[MATMA][BF4], 95% for P[VBTMA][BF4].
The gas sorption of the polymers was measured using a Cahn 1000 electrobalance under ambient atmosphere pressure (592.3 mmHg or 0.78 atm, Laramie, Wyoming USA). The sample used was 1.0 g. A detailed description of this apparatus and the experimental procedures are given in the supporting information.†
In the CO2 sorption of wet P[VBTMA][BF4] experiment, wet N2 saturated with moisture was first introduced to the chambers. When the amount of absorbed water reached 13.8 mol%, N2 was changed to dry CO2, and the CO2 absorption was recorded.
We thank the State of Wyoming (EORI) and the University of Wyoming for financial support. We thank Dr. Jeff Yager for his help with DSC measurements.
Notes and references
- M. J. Earle and K. R. Seddon, ACS Symp. Ser., 2002, 819, 10–25 CAS.
- L. A. Blanchard, D. Hancu, E. J. Beckman and J. F. Brennecke, Nature, 1999, 399, 28–29 CrossRef.
- L. A. Blanchard, Z. Y. Gu and J. F. Brennecke, J. Phys. Chem. B, 2001, 105, 2437–2444 CrossRef CAS.
- L. A. Blanchard and J. F. Brennecke, Ind. Eng. Chem. Res., 2001, 40, 287–292 CrossRef CAS.
- J. L. Anthony, E. J. Maginn and J. F. Brennecke, J. Phys. Chem. B, 2002, 106, 7315–7320 CrossRef CAS.
- A. P.-S. Kamps, D. Tuma, J. Xia and G. J. Maurer, Chem. Eng. Data, 2003, 48, 746–749 Search PubMed.
- P. Husson-Borg, V. Majer and M. F. Costa Gomes, J. Chem. Eng. Data, 2003, 48, 480–485 CrossRef CAS.
- C. Cadena, J. L. S. J. K. Anthony, T. I. Morrow, J. F. Brennecke and E. J. Maginn, J. Am. Chem. Soc., 2004, 126, 5300–5308 CrossRef.
- P. Scovazzo, D. Camper, J. Kieft, J. Poshusta, C. Koval and R. D. Noble, Ind. Eng. Chem. Res., 2004, 43, 6855–6860 CrossRef CAS.
- D. Camper, P. Scovazzo, C. Koval and R. D. Noble, Ind. Eng. Chem. Res., 2004, 43, 3049–3054 CrossRef CAS.
- R. E. Baltus, B. H. Culbertson, S. Dai, H. Luo and D. W. DePaoli, J. Phys. Chem. B, 2004, 108, 721–727 CrossRef CAS.
- E. D. Bates, R. D. Mayton, I. Ntai, Jr. and J. H. Davis, J. Am. Chem. Soc., 2002, 124, 926 CrossRef CAS.
- P. Scovazzo, J. Kieft, D. A. Finan, C. Koval, D. L. DuBois and R. D. Noble, J. Membr. Sci., 2004, 238, 57–63 CrossRef CAS; P. Scovazzo, A. E. Visser, J. H., Jr. Davis, R. D. Rogers, C. A. Koval, D. L. DuBois and R. D. Noble, ACS Symp. Ser., 2002, 818, 69–87 CAS.
- M. D. Macedonia, D. D. Moore and E. J. Maginn, Langmuir, 2000, 16, 3823–3834 CrossRef CAS.
- S. Kato, Y. Tsujita, H. Yoshimizu, T. Kinoshita and J. S. Higgins, Polymer, 1997, 38, 2807–2811 CrossRef CAS.
- Z. Mogri and D. R. Paul, Polymer, 2001, 42, 7781–7789 CrossRef CAS.
- J. L. Budzien, D. McCoy, D. H. Weinkauf, R. A. LaViolette and E. S. Peterson, Macromolecules, 1998, 31, 3368–3371 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: experimental details. See http://www.rsc.org/suppdata/cc/b5/b501940k/ |
| ‡ Visiting Ph.D. student to the University of Wyoming. |
| This journal is © The Royal Society of Chemistry 2005 |