Plausible origins of homochirality in the amino acid catalyzed neogenesis of carbohydrates†
Armando
Córdova
*,
Magnus
Engqvist
,
Ismail
Ibrahem
,
Jesús
Casas
and
Henrik
Sundén
Department of Organic Chemistry, Arrhenius Laboratory, Stockholm University, Sweden. E-mail: acordova1a@netscape.net; acordova@organ.su.se; Fax: + 46 8 15 49 08; Tel: +46 8 162479
First published on 1st March 2005
Abstract
The intrinsic ability of amino acids to catalyze the asymmetric formation of carbohydrates, which enzymes have mediated for millions of years, with significant amplification of enantiomeric excess suggests a plausible ancient catalytic process for the evolution of homochirality.
The origins of the homochirality of natural amino acids and sugars have intrigued researchers for decades.1 The theoretical basis for the evolution of high asymmetry from a diminutive imbalance of enantiomers was suggested more than half a century ago.2 The autocatalytic amplification of enantiomeric excess of a chiral molecule was experimentally demonstrated by Soai and co-workers in the treatment of pyrimidine-5-carboxaldehyde with diisopropylzinc.3 The reaction serves as a mechanistic model for the chemical evolution of homochirality, however, the zinc addition chemistry involved in this transformation is unlikely to have occurred in a prebiotic environment. Extraterrestrial amino acids have been found on carbonaceous meteorites with enantiomeric excesses of up to 9%.4 This investigation has led to the speculation of amino acid catalysis as a potential route for the evolution of biological homochirality.5–7 Amino acids with low optical purity may have initiated the asymmetric neogenesis of hexose carbohydrates, which natural enzymes have catalyzed for millions of years.8 The asymmetric amplification of sugars is directly connected to the evolution of homochiral RNA synthesis as well as to the chiral-selective amino acylation,9,10 which is the first step in the asymmetric protein synthesis. In this context, it would be of paramount interest to determine whether chiral amplification occurs in the plausible amino acid-catalyzed asymmetric formation of hexose carbohydrates.
We recently found that amino acids catalyze the incorporation of molecular oxygen into carbonyl compounds and furnish glycolaldehydes.7 The derived glycolaldehydes are also substrates for the amino acid-catalyzed formation of tetroses under prebiotic conditions.5,11 In addition, we found that amino acids catalyze the asymmetric neogenesis of natural hexose sugars.12
With these results in hand we were intrigued by the possibility that non enantiomerically pure amino acids may have set the seed for the evolution of homochirality of carbohydrates.12–15 Herein, we report that amino acids catalyze the unprecedented formation of hexose carbohydrates with remarkably high amplification of enantiomeric excess. The intrinsic ability of amino acids to catalyze the asymmetric neogenesis of carbohydrates provides a mechanism by which a small initial imbalance in chirality is significantly amplified. The hydroxyproline and proline-catalyzed one-step synthesis of erythrose 1 and allose 2 was selected as the model system (eqn. 1).12,15
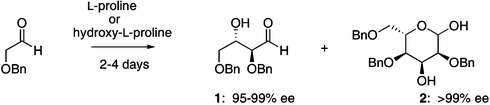 | (1) |
Hence, α-benzyloxy acetaldehyde (1 mmol) was treated with a catalytic amount of proline (10 mol%) in DMF (1 mL). After 2 days the reaction was quenched and the corresponding 2,4,6-tri-O-benzyl allose 2 was isolated by silica-gel column chromatography. Next, allose 2 was converted to the corresponding peracetylated sugar and the enantiomeric excess (ee) was determined. The reaction performed with enantiomerically pure L-proline (> 99% ee) furnished the L-sugar 2 as a predominant diastereomer with > 99% ee. Next, the one-step amino acid-catalyzed asymmetric synthesis of allose 2 was examined using enantio-enriched L-proline as the catalyst (Fig. 1).
 | ||
| Fig. 1 Relation between the enantiomeric excess (ee) of L-proline and that of the newly formed sugar 2 in the one-step catalytic asymmetric synthesis of 2. | ||
Initially, proline with an ee of 80% was used as the catalyst and allose 2 was furnished with > 99% ee. The ee of the furnished sugar 2 did not significantly decrease until the optical purity of the catalyst was below 30% ee. For example, the reaction mediated by proline with 20% ee furnished sugar 2 with 55% ee. Hence, the amino acid-catalyzed formation of the hexose exhibited a significant non-linear effect.6a,16–18 In fact, this is the largest permanent non-linear effect observed for a proline-catalyzed reaction. The reaction catalyzed by proline with 10% ee, which is similar to the optical purity (9% ee) of the extraterrestrial amino acids found on the Murchison meteroite,4 furnished the hexose sugar with a significant augmented enantiomeric excess (33% ee). Thus, amino acids with a low optical purity may have initiated and set the seed of homochirality of sugars. We also performed the reaction utilizing D-proline (> 99 ee) as the catalyst and isolated the opposite enantiomer of the sugar ent-2 with > 99% ee. In addition, the enantiomeric excess of the allose 2 was below the detection level when racemic D,L-proline was employed as the catalyst. These results demonstrate that the configuration of the amino acid determines the configuration of the sugar 2.
The amino acid-catalyzed formation of hexoses is a sequential one-pot direct catalytic asymmetric aldol reaction (Fig. 2), where the initially formed erythrose derivative undergoes a second enamine-catalyzed aldol reaction to yield the sugar with excellent diastereo- and enantioselectivity. The stereoselectivity of the erythrose is set in the first aldol addition step and dictated by the catalytic enamine intermediate, which is formed between the amino acid and the glycoladehyde donor.12–15,18 Next, the erythrose adds to the re-face of the catalytic enamine intermediate via transition state I and allose is formed. The rate of the second anti-selective aldol reaction is significantly slower than the initial aldol addition, which is established by the higher amount of erythrose intermediate as compared to allose. Hence, the hexose formation is the rate-determining step. Furthermore, the tetrose intermediate is less stable than the final hexose adduct.19 We therefore believe that the interaction between non-enantiomerically pure proline and the tetrose intermediate is important for the amplification of asymmetry in the sequential aldol additions.
 | ||
| Fig. 2 The amino acid-catalyzed asymmetric one-step synthesis of allose 2 and the plausible T.S. I of the second aldol addition step. | ||
To establish whether proline is able to discriminate between the two enantiomers of anti-β-hydroxy-aldehydes in sequential cross-aldol reactions,12 we investigated the proline-catalyzed anti-selective propionaldehyde addition to racemic anti-β-hydroxy aldehydes (Scheme 1a). For example, propionaldehyde was reacted with racemic aldol adduct 3 in the presence of a catalytic amount of D-proline. The reaction proceeded with excellent selectivity and polyketide hexose 4 was isolated in 22% yield with > 19 ∶ 1 dr and 95% ee together with remaining acceptor aldehyde 3 in 72% yield with < 5% ee. Hence, D-proline catalyzed a plausible dynamic kinetic resolution and reacted faster with one of the two enantiomers (Scheme 1b). The high selectivity of this transformation indicates that significant enhancement of enantiomeric excess of natural hexoses was present in the second amino acid-catalyzed aldol addition step.
 | ||
| Scheme 1 (a) A two-step synthesis of a polyketide hexose. The first step is performed with racemic proline and the next step with enantiomerically pure D-proline. (b) The potential mechanism of the D-proline catalyzed anti-selective aldol reaction step. | ||
In a prebiotic scenario, the amino acids plausibly catalyzed the formation of both homochiral tetroses and hexoses according to the routes presented herein.5 The initial transfer of asymmetry to the tetrose sugars may have been significantly amplified to the more stable hexose sugars. This mechanism would also have led to the development of different sugar diastereomers. Both tetroses and hexoses have been suggested as building blocks of ancient RNA.20 The amino acid-catalyzed amplification of asymmetry to sugars might potentially have been further amplified to and by ancient RNA.9,10
In summary, it seems conceivable that the amino acid-catalyzed formation of hexoses presented herein may be an example of the theoretical basis for the evolution of homochirality of sugars from a low optical purity of the catalysts. The intrinsic ability of amino acids to catalyze the asymmetric formation of carbohydrates, which enzymes have mediated for millions of years, suggests a plausible ancient catalytic process that perhaps still occurs both on Earth and elsewhere in the universe. Further kinetic and molecular modelling studies are ongoing.
We gratefully acknowledge the Swedish National Research Council and the Wenner-Gren Foundation for financial support. We also thank Prof. Hans Adolfsson and Prof. Jan.-E. Bäckvall for valuable discussions.
Notes and references
- W. A. Bonner, Orig. Life Evol. Biosphere, 1991, 21, 59 CAS; J. L. Bada, Nature, 1995, 374, 594 CrossRef CAS; S. F. Mason, Nature, 1985, 314, 400 CrossRef.
- F. C. Frank, Biochem. Biophys. Acta, 1953, 11, 459 CrossRef CAS.
- K. Soai, T. Shibata, H. Morioka and K. Choji, Nature, 1995, 378, 767 CrossRef CAS; T. Shibata, H. Morioka, T. Hayase, K. Choji and K. Soai, J. Am. Chem. Soc., 1996, 118, 471 CrossRef CAS.
- J. R. Cronin and S. Pizzarello, Science, 1997, 275, 951 CrossRef CAS.
- S. Pizzarello and A. L. Weber, Science, 2004, 303, 1151 CrossRef CAS.
- For an excellent study by Blackmond and co-workers see: (a) S. P. Methew, H. Iwamura and D. Blackmond, Angew. Chem., Int. Ed., 2004, 43, 3317 CrossRef CAS; (b) For asymmetric amplification in a polypeptide catalyzed epoxidation see: D. R. Kelly, A. Meed and S. M. Roberts, Chem. Commun., 2004, 2021 Search PubMed.
- A. Córdova, H. Sundén, M. Enqvist, I. Ibrahem and J. Casas, J. Am. Chem. Soc., 2004, 126, 8914 CrossRef CAS.
- J. M. Berg, J. L. Tymoczko and L. Stryer, Biochemistry, W. H. Freeman & Co., New York, 2002 Search PubMed.
- G. F. Joyce, G. M. Visser, C. A. van Boeckel, J. H. van Boom, L. E. Orgel and J. van Westrenen, Nature, 1984, 310, 602 CrossRef CAS.
- K. Tamura and P. Schimmel, Science, 2004, 305, 1253 CrossRef CAS.
- For example, L-valine, L-alanine and L-phenylalanine catalyzed the dimerization of glycol aldehyde in water to furnish D-threose with 12–15% ee. L-serine and L-proline furnished L-threose with 6 and 9% ee, respectively. For a Zn–amino acid mediated sugar synthesis under prebiotic conditions see: J. Kofoed, M. Machuqueiro, J.-L. Reymond and T. Darbre, Chem. Commun., 2004, 26, 1540 Search PubMed.
- J. Casas, M. Enqvist, I. Ibrahem, B. Kaynak and A. Córdova, Angew. Chem., Int. Ed., 2005, 44, 1343 CrossRef.
- For excellent reviews see: P. I. Dalko and L. Moisan, Angew. Chem., Int. Ed., 2001, 40, 3726 Search PubMed; B. List, Tetrahedron, 2002, 58, 5573 CrossRef CAS; P. I. Dalko and L. Moisan, Angew. Chem., Int. Ed., 2004, 43, 5138 CrossRef CAS.
- For the proline-catalyzed asymmetric aldol reactions see: (a) B. List, R. A. Lerner and C. F. Barbas, III, J. Am. Chem. Soc., 2000, 122, 2395 CrossRef CAS; (b) W. Notz and B. List, J. Am. Chem. Soc., 2000, 122, 7386 CrossRef CAS; (c) Z. G. Hajos and D. R. Parrish, J. Org. Chem., 1974, 39, 1615 CrossRef CAS; (d) U. Eder, R. Sauer and R. Wiechert, Angew. Chem., Int. Ed., 1971, 10, 496 CrossRef. For proline-catalyzed asymmetric cross-aldol reactions see: A. B. Northrup and D. W. C. MacMillan, J. Am. Chem. Soc., 2002, 124, 6798 Search PubMed; A. B. Northrup, I. K. Mangion, F. Hettche and D. W. C. MacMillan, Angew. Chem., Int. Ed., 2004, 43, 2152 Search PubMed.
- It was previously believed that proline does not catalyze the formation of natural hexoses, see: A. B. Northrup and D. W. C. MacMillan, Science, 2004, 305, 1752 Search PubMed; E. J. Sorensen and G. M. Sammis, Science, 2004, 305, 1725 CrossRef CAS . At present we do not know why the hexoses were not observed.
- A. Córdova, H. Sundén, A. Bøgevig, M. Johansson and F. Himo, Chem. Eur. J., 2004, 10, 3673 CrossRef CAS.
- Proline-catalyzed intramolecular aldol reactions were believed to exhibit non-linear effects, see: (a) C. Puchot, O. Samuel, E. S. Duñach, S.-H. Zhao, C. Agami and H. B. Kagan, J. Am. Chem. Soc., 1986, 103, 2353 CrossRef; (b) D. Guillaneux, S.-H. Zhao, O. Samuel, D. Rainford and K. B. Kagan, J. Am. Chem. Soc., 1994, 116, 9430 CrossRef CAS.
- Recent studies demonstrate a linear relationship for the asymmetric intramolecular and intermolecular aldol reactions. L. Hoang, S. Bahmanyar, K. N. Houk and B. List, J. Am. Chem. Soc., 2003, 125, 16 Search PubMed.
- The ee of the erythrose derivative decreased with increased reaction time. This was not observed for the hexose 1. For the instability of tetroses in water see: G. Springsteen and G. F. Joyce, J. Am. Chem. Soc., 2004, 126, 9578 Search PubMed.
- L. E. Orgel, Science, 2000, 290, 1306 CrossRef CAS; N. Hall, Chem. Commun., 2004, 1247 RSC and references cited therein.
Footnote |
| † Electronic supplementary information (ESI) available: experimental procedures. See http://www.rsc.org/suppdata/cc/b5/b500589b/ |
| This journal is © The Royal Society of Chemistry 2005 |