Killing three birds with one stone—simultaneous operando EPR/UV-vis/Raman spectroscopy for monitoring catalytic reactions
Angelika
Brückner
Institut für Angewandte Chemie Berlin-Adlershof†, P.O. Box 961156, D-12474 Berlin, Germany. E-mail: brueckner@aca-berlin.de; Fax: +49 (0)30 6392 4454; Tel: +49 (0)30 6392 4301
First published on 4th February 2005
Abstract
For the first time, three operando methods, namely EPR, UV-vis and laser-Raman spectroscopy have been applied in parallel on the same V/TiO2 catalyst and under identical reaction conditions during oxidative dehydrogenation of propane to elucidate structure–reactivity relationships in status operandi.
A recent trend of growing importance in catalysis research comprises the monitoring of catalysts under reaction conditions, including analysis of product composition, i.e., in status operandi, by more than one operando technique at the same time and in the same reactor. The crucial advantage of such multiple couplings is the fact that results are obtained from the very same catalyst sample for which identical reaction conditions are guaranteed. Thus, the spectroscopic responses from different methods can be directly interrelated which may provide more comprehensive and more relevant results. This is not possible without doubt when, as usual so far, the experiments are performed with different samples (though from the same batch) in different reaction cells, since identical reaction conditions, in particular, with respect to reactant flow and temperature distribution within the samples are not ensured. This communication illustrates the special benefits obtained by using operando EPR/UV-vis-DRS/laser-Raman spectroscopy—the first threefold coupling of operando techniques—for analysis of structure–function relationships during the oxidative dehydrogenation of propane (ODP).
For performing such triple operando experiments, the previously established set-up for simultaneous operando EPR/UV-vis spectroscopy consisting of a fibre optical sensor implemented into an EPR fixed-bed flow reactor1 has been extended by focusing the beam of a 785 nm diode laser of a Kaiser Optical Systems RXN spectrometer through a front hole in the EPR cavity onto the catalyst bed within the reactor. Different from the previous set-up,1 heat was transferred to the reactor by a stream of N2 which was pre-heated by passing over an electrical heater. Heater power and N2 flow rate were controlled by a Bruker variable temperature control unit. Details of spectra acquisition have been described elsewhere,1 however, UV-vis spectra in the present work were recorded by an AVASPEC fibre optical spectrometer (Avantes) and a 200 × 1.5 mm quartz sensor (Optran UV 1500/1800 T, CeramOptec GmbH). Raman spectra were recorded with a laser power of 25 mW and an acquisition time of 3 s. Five scans were accumulated for each spectrum. Computer simulation of EPR spectra was performed with the program SIM14S of Lozos et al.2 using the spin Hamiltonian
| H = μBSgB0 + SAI | (1) |
A 6 wt% V/TiO2 catalyst was used which was prepared by thermal spreading of V2O5 on a commercial anatase carrier (80 m2 g−1, 2.2% sulfate, Millennium Chemicals). The appropriate amount of V2O5 was mixed with the support by intense grinding in an agate mortar followed by 2 h calcination in an air flow at 600 °C. This catalyst was chosen for two reasons: (i) it contains vanadium in different forms ranging from isolated, via oligomeric V surface species, to V2O5 microcrystals and thus allows conclusions to be derived on the reaction-dependent behavior of these different species simultaneously and (ii) V/TiO2 showed superior activity in comparison to VOx on other support materials.3,4 Therefore it was concluded that the V–O–Ti moiety might be the active species but no direct experimental insight was given.5 From the new operando EPR/UV-vis/Raman spectroscopy setup, direct evidence for the participation of TiO2 is expected.
For operando experiments, 60 mg catalyst particles (250–355 µm) were pretreated for 30 min in an air flow at 450 °C and cooled to room temperature. Then, the catalyst was heated stepwise in a mixture of 8.3% C3H8, 8.3% O2/N2 (total flow of 24 ml min−1) to different temperatures with 10 min isothermal hold at each temperature for recording the spectra and GC analyses. The reactant gas flow was mixed by a gas dosing system containing mass flow controllers (Bronckhorst). For online product analysis, the reactor outlet was connected to a GC 17AAF capillary gas chromatograph (Shimadzu) equipped with a FID and a 30 m × 0.32 mm Silicaplot column (Chrompack).
At ambient and higher temperatures, EPR is exclusively sensitive for tetravalent vanadium and can detect isolated V4+ sites in distorted octahedral and/or square-pyramidal coordination by characteristic hyperfinestructure signals with axial g and A tensors besides clustered V4+Ox that give rise to a broad isotropic singlet. In contrast, Raman spectra reflect pentavalent V species only, whereby both isolated and polymeric VOx surface species can be distinguished from crystalline V2O5 and TiO2. UV-vis spectroscopy, in general, can visualize both fully oxidized V5+ by intense charge-transfer (CT) bands in the UV range as well as reduced V species by weak d–d transitions in the visible range. However, for the V/TiO2 catalyst used in this work, UV-vis spectroscopy can provide information on V2O5 and reduced V species only, since the CT range of dispersed surface V5+Ox is superimposed by the strong absorption of TiO2. Due to the described specific limitations of each technique, their simultaneous coupling is of unique advantage. In the following, a joint discussion of the results obtained from the three spectroscopic techniques is given, dependent on the reaction conditions.
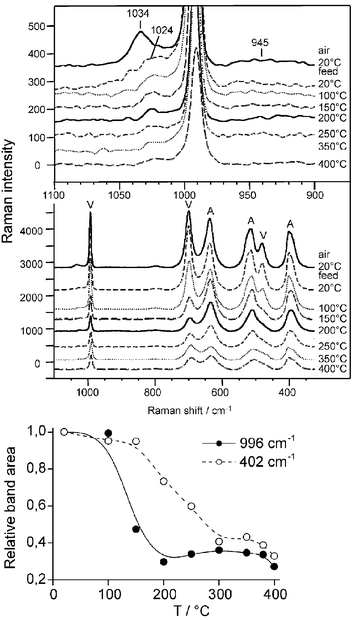
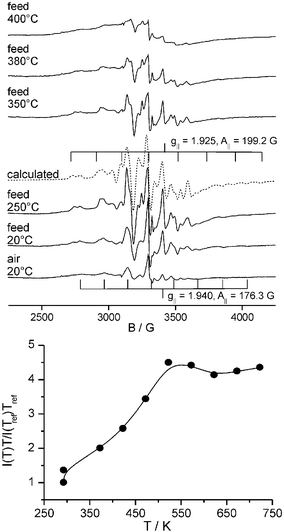
The Raman spectrum of V/TiO2 after oxidative pretreatment shows bands of crystalline anatase (marked by A), V2O5 microcrystals (marked by V) as well as of isolated and polymeric O![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) VOx surface species at 1034 cm−1 and 945 cm−1, respectively (Fig. 1).6,7 A further band at 1024 cm−1 also assigned to polymeric surface species7,8 might be hidden by the bands at 1034 and 996 cm−1. In the corresponding EPR spectrum, a signal 1 with hyperfine structure (hfs) (g|| = 1.940, A|| = 176.3 G, derived by spectra simulation) being typical for VO2+ species in anatase9 is detected which arises from V sites persisting oxidative pretreatment (Fig. 2). The related UV-vis spectrum (Fig. 3) shows a broad band around 470 nm arising from a CT transition of V2O5 microcrystals.10 As explained above, CT bands of isolated and low polymerized VOx species falling below 400 nm,10 are obscured by the strong absorption of TiO2.
VOx surface species at 1034 cm−1 and 945 cm−1, respectively (Fig. 1).6,7 A further band at 1024 cm−1 also assigned to polymeric surface species7,8 might be hidden by the bands at 1034 and 996 cm−1. In the corresponding EPR spectrum, a signal 1 with hyperfine structure (hfs) (g|| = 1.940, A|| = 176.3 G, derived by spectra simulation) being typical for VO2+ species in anatase9 is detected which arises from V sites persisting oxidative pretreatment (Fig. 2). The related UV-vis spectrum (Fig. 3) shows a broad band around 470 nm arising from a CT transition of V2O5 microcrystals.10 As explained above, CT bands of isolated and low polymerized VOx species falling below 400 nm,10 are obscured by the strong absorption of TiO2.
 | ||
| Fig. 1 Raman spectra and relative band area at 996 and 402 cm−1. | ||
 | ||
| Fig. 2 EPR spectra and normalized spectral intensity (Tref = 293 K). | ||
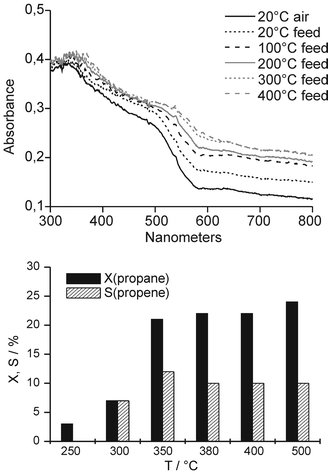
Upon switching to feed at 20 °C, Raman signals of crystalline V2O5 and TiO2 remain unchanged while the band at 1034 cm−1 disappears immediately rendering the band at 1024 cm−1 well visible (Fig. 1). In EPR, hfs signal 1 increases in intensity and a new hfs signal 2 (g|| = 1.925, A|| = 199.2 G) appears from VO2+ species which must have been formed upon reduction of isolated VO3+ sites by propane (Fig. 2). In the Raman spectrum of oxidatively pretreated V/TiO2 (Fig. 1), these initial VO3+ sites are reflected by the band at 1034 cm−1 that disappears upon contact with propane. In the corresponding UV-vis spectrum (Fig. 3), the reduction of VO3+ to VO2+ leads to increasing absorbance in the range of d–d transitions of reduced V species above 500 nm. The direct interconversion of isolated VO3+ (Raman band at 1034 cm−1) into VO2+ (EPR hfs signal 1 and 2, rising light absorption above 500 nm) which is simultaneously confirmed by all three techniques, shows clearly that isolated V5+ species on anatase are already reduced to V4+ at 20 °C. Moreover, the direct relation between the disappearance of the Raman band at 1034 cm−1 and the increase of two VO2+ EPR lines is evidence that this Raman band cannot be exclusively due to tetrahedral OV5+O3 species as assumed so far4,7 but reflects at least two different square-pyramidal, isolated OV5+O4 moieties. This is unique information that could only be obtained by the triple operando approach and confirms both the previous suggestions that the VO group gives rise to vibrations largely independent of the rest of the molecule4,7 as well as the assumption that VOx on anatase might be connected to the support by four oxygen bridges.6
Upon a stepwise increase of temperature, the Raman band of polymeric surface VxOy (1024 cm−1) decreases gradually and those of V2O5 drop to a constant low level above 200 °C (Fig. 1). This is also evident from a plot of the relative band area derived by integration in the range between 900 and 1100 cm−1 (Fig. 1). The Raman bands of anatase loose intensity, too, although more slowly and at higher temperature compared to V2O5. When the V-free TiO2 support is treated under the same conditions, band intensity decreases, too, but to a markedly smaller extent. This suggests that the redox activity of the support might be enhanced by the presence of vanadia species dispersed on its surface.
In the corresponding EPR spectra (Fig. 2), a broad singlet of polymeric V4+Ox species superimposed on the VO2+ hfs signals grows with rising temperature which reflects the reduction of polymeric surface V5+Ox and V2O5 in parallel to the decreasing Raman band at 1024 cm−1 and those marked with V (Fig. 1). In general, EPR signals of paramagnetic species decrease linearly with rising temperature due to the Curie–Weiss law. To eliminate this effect, the normalized intensity function I(T)T/I(Tref)Tref is plotted against the absolute temperature, in which I is the double integral of the total EPR signal and Tref is a reference temperature (this work: 293 K) (Fig. 2). From this plot it is evident that the amount of V4+ increases until it reaches a constant value at ≈220 °C. This is in perfect agreement with the decrease of the V5+ Raman signal area between 900 and 1100 cm−1 (Fig. 1). Considering the various EPR subsignals, it appears that hfs signal 2 vanishes gradually above 250 °C. It is also not seen anymore after cooling the used catalyst to room temperature. Most probably, the respective isolated VO2+ sites are reduced to EPR-silent V3+ which might be inactive in the catalytic reaction11 while the isolated VO2+ sites reflected by hfs signal 1 as well as the polymeric V4+ sites comprised by the broad isotropic singlet are not further reduced.
In the corresponding UV-vis spectra (Fig. 3), the increase of light absorption above 500 nm, reflecting reduced V sites, levels off between 200 and 300 °C. This agrees pretty well with EPR and Raman results described above. In general, it is not straightforward to discriminate between the effect of reduced V species and coke deposits since the latter give rise to light absorption in the same wavelength range.12 However the almost perfect agreement of the absorbance behaviour above 500 nm in the UV-vis spectra (Fig. 3) with the behaviour of the normalized EPR intensity comprising exclusively tetravalent vanadium (Fig. 2) suggests that the UV-vis spectra reflect V reduction only, while coke deposition is negligible.
Propane conversion becomes measurable at 250 °C and increases to a constant level above 350 °C. At this temperature, virtually all monomeric and polymeric OV5+Ox surface species and a considerable part of the V2O5 crystals are reduced, however, this does not suppress propane conversion. Moreover, propene selectivity increases, too, with rising extent of V5+ reduction. In contrast to this, it has been claimed previously that propene selectivity is favoured by V5+ surface species and V2O5 microcrystals are not active in ODP.13
In summary, the above presented results illustrate that more authentic information and new insight into a well-known catalytic system such as V/TiO2 during ODP can be obtained when simultaneous operando EPR/UV-vis/Raman spectroscopy is used instead of the respective separate operando techniques, whereby it must be stressed, that even separate operando EPR, UV-vis and Raman measurements have not been performed so far on the same V/TiO2 catalyst during ODP. Thus, it could be shown that isolated VO2+ sites in slightly different octahedral and/or square-pyramidal coordination are reacting first followed by polymerized VOx surface species and V2O5 crystals. The reactivity of those species has been controversially discussed so far, and highly dispersed VOx surface species with more than four oxygen ligands were rarely considered to be involved in the reaction. Moreover, this work provides clear evidence that different V4+ species (isolated and polymeric VO2+ in direct contact with TiO2 as well as VO2+ sites on the surface of vanadia microcrystals) dominate under the reaction conditions and are likely active sites. Finally, the participation of the TiO2 support in the redox process has been directly shown. The onset of propane conversion at rather low temperature is most probably due to the promotion of the catalyst redox activity by participation of TiO2.
The author thanks the Federal Ministry for Education and Research of Germany, the EU and the Federal State of Berlin (Dept. for Science, Research and Culture) for financial support.
Notes and references
- A. Brückner, Chem. Commun., 2001, 2122 RSC.
- G. P. Lozos, B. M. Hofman and C. G. Franz, Quantum Chemistry Program Exchange, 1973, no. 265 Search PubMed.
- M. A. Bañares, M. Martinez-Huerta, X. Gao, I. E. Wachs and J. L. G. Fierro, Stud. Surf. Sci. Catal., 2000, 130, 3125.
- I. E. Wachs, J.-M. Jehng, G. Deo, B. M. Weckhuysen, V. Guliants and J. B. Benziger, Catal. Today, 1996, 32, 47 CrossRef CAS.
- M. A. Bañares and I. E. Wachs, J. Raman Spectrosc., 2002, 33, 359 CrossRef CAS.
- G. T. Wendt, L.-J. Leu and A. T. Bell, J. Catal., 1992, 134, 479 CrossRef CAS.
- L. J. Burcham, G. Deo, X. Gao and I. E. Wachs, Top. Catal., 2000, 11(12), 85 CrossRef.
- G. G. Cortez and M. A. Bañares, J. Catal., 2002, 209, 197 CrossRef.
- O. B. Lapina, A. A. Shubin, A. V. Nosov, E. Bosh, J. Spengler and H. Knözinger, J. Phys. Chem. B, 1999, 103, 7599 CrossRef CAS.
- G. Centi, S. Perathoner, F. Trifiró, A. Aboukais, C. F. Aissi and M. Guelton, J. Phys. Chem., 1992, 96, 2617 CrossRef CAS.
- U. Bentrup, A. Brückner, C. Rüdinger and H.-J. Eberle, Appl. Catal., A, 2004, 269, 237 CrossRef CAS.
- A. Brückner, Phys. Chem. Chem. Phys., 2003, 5, 4461 RSC.
- X. Gao, J.-M. Jehng and I. E. Wachs, J. Catal., 2002, 209, 43 CrossRef CAS.
Footnote |
| † A member of the EU funded Coordination Action of Nanostructured Catalytic Oxide Research and Development in Europe (CONCORDE) |
| This journal is © The Royal Society of Chemistry 2005 |