Stereochemistry of the reaction catalysed by 2-hydroxy-6-keto-6-phenyl-hexa-2,4-dienoic acid 5,6-hydrolase (BphD)
Jian-Jun
Li
and
Timothy D. H.
Bugg
*
Department of Chemistry, University of Warwick, Coventry, CV4 7AL, UK. E-mail: T.D.Bugg@warwick.ac.uk; Fax: 02476-524112; Tel: 02476-573018
First published on 26th November 2004
Abstract
The stereochemical course of the reaction catalysed by C-C hydrolase BphD from Burkholderia xenovorans LB400 occurs with replacement of a benzoyl group by hydrogen with overall retention of stereochemistry.
C-C hydrolase enzymes catalyse the hydrolytic cleavage of a carbon–carbon bond adjacent to a ketone, yielding a carboxylic acid product.1 A class of C-C hydrolases are found on bacterial meta-cleavage pathways responsible for the degradation of aromatic compounds in soil.2 Amino acid sequence alignments of these enzymes,3 and structure determination of hydrolase BphD from Rhodococcus RHA1,4 have revealed that they are members of the αβ-hydrolase family, containing a Ser-His-Asp catalytic triad.
Mechanistic studies of C-C hydrolase MhpC, on the phenylpropionic acid catabolic pathway of Escherichia coli, have established that the reaction occurs with enzymatic insertion of the H-5E hydrogen of the product 2-hydroxypentadienoic acid, and replacement of a succinyl group with overall retention of stereochemistry.5,6 Pre-steady state kinetic analysis of MhpC has provided evidence for a keto-intermediate,7 which is attacked by water via a general base-catalysed mechanism.8 We have recently established a synthetic route to the aryl meta-ring fission intermediates on the biphenyl catabolic pathway, responsible for the degradation of polychlorinated biphenyls.9 The availability of synthetic intermediates on this pathway enables mechanistic studies of C-C hydrolase BphD, which bears 50% amino acid sequence identity to E. coli MhpC. Here we report the elucidation of the stereochemistry of the BphD-catalysed reaction (Fig. 1), and pre-steady state kinetic analysis.
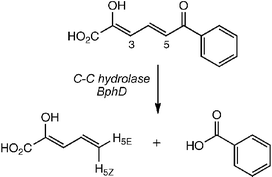 | ||
| Fig. 1 Reaction catalysed by C-C hydrolase BphD. H-3 and H-5 exchanged with 2H prior to reaction in 1H2O. | ||
2-Hydroxy-6-keto-6-phenyl-hexa-2,4-dienoic acid was synthesised by Heck coupling of 1-phenyl-prop-2-en-1-ol and ethyl 3-bromo-2-acetoxy-propenoate.9 C-C hydrolase from Burkholderia xenovorans LB40010,11 was expressed as an N-terminal His6 fusion protein, and purified to near homogeneity (specific activity 11.8 u/mg). Steady-state kinetic parameters for (His)6-BphD were determined to be KM 2.0 µM and kcat 6.5 s−1, similar to reported values for native BphD.11 Enzymatic conversion in 50 mM potassium phosphate buffer, pH 8.0, followed by extraction of the unstable 2-hydroxypentadienoic acid product (t1/2 5–10 min12), gave the 1H NMR spectrum shown in Fig. 2A, identical to that of the MhpC enzymatic product.6 The separation of the H-5E hydrogen (5.42 ppm) and H-5Z hydrogen (5.18 ppm) allows the determination of stereochemistry by 1H NMR spectroscopy. Attempted enzymatic conversions of substrate in 2H2O resulted in very rapid 2H exchange in the substrate dienol, obscuring the stereochemical determination. Therefore, the deuteriated substrate was prepared by exchange of H-5 and H-3 of the dienol substrate, by incubation of substrate in 2H2O. Conversion of 10 mg of the deuteriated substrate in 1H2O by 8 units of BphD, and direct monitoring of the reaction by 1H NMR spectroscopy, gave the spectrum shown in Fig. 2B. Integration of the 1H signals for H-5E (89%) and H-5Z (31%, see Table 1) shows that the H-5E hydrogen is inserted by BphD. There is 31% exchange of the H-5Z hydrogen, similar to that observed for MhpC,6 which is due to partial release of the ketonised reaction intermediate, followed by non-specific re-enolisation in solution. Therefore, the stereochemistry of the BphD-catalysed reaction occurs with replacement of the benzoyl substituent by hydrogen with overall retention of stereochemistry.
 | ||
| Fig. 2 Part of 1H NMR spectra for (A) conversion of 1H-RFP in 1H2O, (B) conversion of 2H-RFP in 1H2O. | ||
| H-4 | H-3 | H-5Z | H-5E | |
|---|---|---|---|---|
| BphD | 1.00 | 0.15 | 0.31 | 0.89 |
| MhpC6 | 1.00 | 0.02 | 0.30 | 1.00 |
In order to examine the kinetic mechanism for the BphD-catalysed reaction, pre-steady state kinetic analysis of the enzyme-catalysed reaction was carried out at 30.3 µM substrate and 30.3 µM BphD in 50 mM potassium phosphate buffer pH 8.0. Observation at 430 nm gave a single exponential curve for substrate consumption (k = 9.4 s−1). Observation of product appearance at 270 nm also gave a single exponential curve (k = 10.2 s−1). Therefore, only a single step kinetic mechanism is observed for BphD, whereas a two-step kinetic mechanism is observed for MhpC, comprising a fast initial keto–enol tautomerisation, followed by rate-limiting C–C cleavage.7 This result implies that the initial ketonisation step is much slower, and rate-limiting, in the BphD catalytic cycle.
In order to probe further the relative energy barriers in the BphD reaction, the solvent kinetic isotope was measured in 100% 2H2O. A value of 1.76 ± 0.02 was measured on vmax, higher than the value of 1.42 measured previously for MhpC.6 The higher solvent kinetic isotope is consistent with the initial tautomerisation step being rate-limiting, since keto–enol tautomerisation involves proton transfer with an active site base. The slower tautomerisation by BphD may reflect the greater resonance stabilisation of the aryl substrate for BphD, or a lesser degree of substrate destabilisation than in MhpC.7
The availability of a crystal structure for BphD from Rhodococcus,4 together with these mechanistic data, allows us to propose a more detailed catalytic mechanism for the BphD-catalysed reaction, shown in Fig. 3. The substrate is predicted to bind with the C-1 carboxylate interacting with Arg-188 at the bottom of the active site, and with the C-6 carbonyl positioned between (and beneath) the sidechains of His-263 and Ser-110. Our previous observation that BphD is able to process a reduced substrate containing a secondary alcohol at C-6,13 together with mechanistic studies on C-C hydrolase MhpC,6–8 implies that C–C cleavage proceeds via a general base mechanism, not a nucleophilic mechanism. His-263 appears to be responsible for both keto–enol tautomerisation and deprotonation of the catalytic water molecule, since there are no other acid–base residues in the vicinity of the active site.4
 | ||
| Fig. 3 Proposed catalytic mechanism for C-C hydrolase BphD, illustrating the reaction stereochemistry. | ||
There are two stereochemical mechanisms that could give rise to insertion of the H-5E hydrogen: either protonation at the C-5 proS hydrogen, followed by C–C cleavage onto the re face of the 3,4-double bond; or protonation at the C-5 proR hydrogen, followed by C–C cleavage onto the si face.5 The orientation of the bound substrate, in relation to Ser-110 and His-263, requires that the scissile C5–C6 bond must rotate towards Ser-110, and therefore that C–C cleavage occurs onto the re face. Twisting of the dienol substrate towards Ser-110 would facilitate protonation at C-5 at the proS hydrogen by His-263, hence the mechanism shown in Fig. 3 would result in the observed labelling of the H-5E position.
In summary, we have found that the stereochemistry of the reaction catalysed by C-C hydrolase BphD occurs with insertion of the H-5E hydrogen, and overall replacement of a benzoyl group by hydrogen with retention of stereochemistry. This is the same stereochemical course as C-C hydrolase MhpC from E. coli, but the two enzymes show different kinetic behaviour under single turnover conditions, implying that keto–enol tautomerisation is rate-limiting in this enzyme. The availability of a synthetic route to the BphD substrate will allow a more detailed examination of the catalytic roles of His-263 and Ser-110 in this enzyme, which will be reported in due course.
This work was supported by BBSRC (grant B20467). We thank Dr Sharon Mendel and Chen Li (University of Warwick) for assistance with molecular biology and stopped flow kinetics.
Notes and references
- D. Pokorny, W. Steiner and D. W. Ribbons, Trends Biotechnol., 1997, 15, 291–296 CrossRef.
- T. D. H. Bugg and C. J. Winfield, Nat. Prod. Rep., 1998, 15, 513–530 RSC.
- E. Diaz and K. N. Timmis, J. Biol. Chem., 1995, 270, 6403–6411 CrossRef CAS.
- N. Nandhagopal, A. Yamada, T. Hatta, E. Masai, M. Fukuda, Y. Mitsui and T. Senda, J. Mol. Biol., 2001, 309, 1139–1151 CrossRef CAS.
- W. W. Y. Lam and T. D. H. Bugg, J. Chem. Soc., Chem. Commun., 1994, 1163–1164 RSC.
- W. W. Y. Lam and T. D. H. Bugg, Biochemistry, 1997, 36, 12242–12251 CrossRef CAS.
- I. M. J. Henderson and T. D. H. Bugg, Biochemistry, 1997, 36, 12252–12258 CrossRef CAS.
- S. M. Fleming, T. A. Robertson, G. J. Langley and T. D. H. Bugg, Biochemistry, 2000, 39, 1522–1531 CrossRef CAS.
- D. M. Speare, P. Olf and T. D. H. Bugg, Chem. Commun., 2002, 2304–2305 RSC.
- B. Hofer, S. Backhaus and K. N. Timmis, Gene, 1994, 144, 9–16 CrossRef CAS.
- S. Y. K. Seah, G. Terracina, J. T. Bolin, P. Riebel, V. Snieckus and L. D. Eltis, J. Biol. Chem., 1998, 274, 22943–22949 CrossRef CAS.
- J. R. Pollard, I. M. J. Henderson and T. D. H. Bugg, Chem. Commun., 1997, 1885–1886 RSC.
- D. M. Speare, S. M. Fleming, M. N. Beckett, J. J. Li and T. D. H. Bugg, Org. Biomol. Chem., 2004, 2, 2942–2950 RSC.
| This journal is © The Royal Society of Chemistry 2005 |