Microreactor-based reaction optimization in organic chemistry—glycosylation as a challenge†
Daniel M.
Ratner‡
a,
Edward R.
Murphy‡
b,
Manish
Jhunjhunwala
b,
Daniel A.
Snyder
a,
Klavs F.
Jensen
*b and
Peter H.
Seeberger
*ac
aDepartment of Chemistry, Massachusetts Institute of Technology, Cambridge, MA, USA
bDepartment of Chemical Engineering, MIT, 77 Massachusetts, Avenue, Cambridge, MA 02139, USA. E-mail: kfjensen@mit.edu.; Fax: 617-258-8224; Tel: 617-253-4589
cLaboratorium für Organische Chemie, ETH Hönggerberg, HCI F315, Wolfgang-Pauli-Str. 10, CH-8093, Zurich, Switzerland. E-mail: seeberger@org.chem.ethz.ch; Fax: 41 1 633 12 35; Tel: 41 1 633 21 03
First published on 17th December 2004
Abstract
Glycosylation reactions are performed rapidly over a wide range of conditions as an example of microreactor-based method optimization and process development in organic chemistry.
Although glycosylations have been carried out for more than a century, the union of glycosylating agent and nucleophile to form a glycosidic linkage is a notoriously difficult undertaking.1 Glycoside formation depends on the conformation, sterics, and electronics of both reaction partners. The challenge in accurately predicting the reactivity of the coupling partners makes it difficult to foresee the outcome of the reaction. In addition, reaction variables such as concentration, stoichiometry, temperature, reaction time, and activator play an indisputable role in the outcome of a given glycosylation.2 This complexity is shared by many other organic transformations in which multiple factors determine the outcome of the reaction. In both academic and industrial settings, much of the effort spent by synthetic organic chemists is consumed searching for optimal reaction conditions to achieve a particular transformation. Method optimization frequently requires the commitment of time and large quantities of valuable starting materials.3 The ability to find ideal reaction conditions quickly and efficiently therefore has a major impact on the practice and pace of research and development in organic chemistry.
Microfluidic-based devices are capable of performing a wide range of single and multiphase organic reactions.4 In addition to requiring small quantities of reagent, submillimeter reaction channels allow for the precise control of reaction variables, such as reagent mixing, flow rates, reaction time, and heat and mass transfer. Microfluidic devices are also amenable to integrated reaction monitoring, using UV/VIS, IR, NMR, mass spectrometry (MS), and LC/MS.5 Unlike conventional bench-top batch reactions, microreactors are easily scalable, rendering a device capable of both analytical and semi-preparative scales of production. Finally, the microreactor format is amenable to automation of reaction optimization.
Here, we use continuous flow microreactors to systematically study the glycosylation reaction as an example of a challenging organic transformation. Optimization of yield and the selection of optimal reaction time and temperature is the goal, in addition to gaining an understanding of the formation of different side products. The five-port silicon microreactor (Fig. 1a) was designed with three primary inlets to mix and react glycosylating agent, nucleophile (acceptor), and activator. In order to ensure adequate mixing and long residence times, the reactor is split into a mixing and a reaction zone. Once mixed, the reactants enter a reaction zone which is terminated by a secondary inlet used to quench the reaction. The quenched reaction stream then exits the reactor for collection and analysis (Fig. 1b).
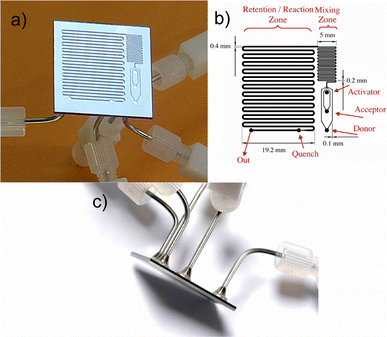 | ||
| Fig. 1 a) Silicon microfluidic microreactor. b) Schematic of microreactor system, comprised of three primary inlets, a mixing and reaction zone, a secondary inlet for quench, and an outlet for analysis/collection. c) Soldered joints of microreactor, also perspective of device from side. | ||
Microfluidic channels were etched into a silicon wafer and capped by a Pyrex wafer via an anodic bond. This construction was chosen for its compatibility with a wide range of chemical reagents, as well as the high thermal conductivity of silicon—facilitating rapid thermal equilibration and temperature control.6 Moreover, the silicon can be oxidized to create a glass surface throughout the resulting microchannels. Deep reactive ion etching techniques (DRIE)7 make it easier to realize deep aspect ratio structures in silicon than glass. Thus, the use of DRIE and subsequent oxidation and anodic bonding to pyrex facilitates making microreactors with glass surface properties. All ports were directly connected by soldering joint to stainless steel tubing to allow the use of solvents, such as CH2Cl2.
An UV-active compound (methyl 2,3,4,6-tetra-O-benzyl-α-D-mannopyranoside)8 was used as an HPLC standard and added to the quench syringe which enters through the secondary inlet (after the reaction zone). The standard was selected for high UV/Vis absorbance, and compatibility with the reacting species. The HPLC standard normalizes the output stream for HPLC analysis by compensating for solvent evaporation and variability in the volume of collected sample.
Initially, the microchemical system was used to carry out the mannosylation of diisopropylidene galactose 1 with mannosyl trichloroacetimidate 2 upon activation with 0.2 equivalents TMSOTf in anhydrous dichloromethane. The reaction was quenched with a solution of triethylamine that also contained the HPLC standard (Scheme 1a). Since the concentration of the reagents in the reactor is determined not only by the concentration inside the syringe but also the flow rate of each stream, all flow rates were maintained in proportion to that of the donor inlet stream. The reaction temperature was varied from −78 to 20 °C, with glycosyl donor stream flow rates of 10, 20, 40 and 80 µl min−1, that resulted in reactor residence times (reaction time) of 26.7, 53.4, 106.8, and 213.5 seconds. Glycosylating agent 2 (1.2 equivalents) and nucleophile 1 (1.0 equivalents) were flowed through the microreactor with reaction zone concentrations of 0.0136 M and 0.0114 M respectively. The triethylamine quench contained the HPLC standard (1.0 equivalent) and dichloromethane to increase solubility.
 | ||
| Scheme 1 (a) Sample glycosylation of glycosyl donor 2 and nucleophile (acceptor) 1 to fashion disaccharide 3. Formation of orthoester 4 is also often observed. (b) Glycosylation reaction involving glycosylating agent 2 (mannosyl donor) and nucleophile 5 (acceptor) to fashion disaccharide 6. Formation of orthoester 7 is also often observed. | ||
HPLC analysis of the crude samples, normalized with internal standard, illustrates a clear relationship between reaction temperature, reaction time and formation of product (Fig. 2a). For a given reaction time, the yield of product increases with temperature until maximum conversion is achieved. Correspondingly, at temperatures lower than the optimum, yield increases with increasing reaction time (i.e. decreasing flow rate). Importantly, we were able to observe the formation of orthoester 4 as a major side product at lower temperatures. Frequently encountered in glycosylations involving a C2-O-acetate, the orthoester is observed at low temperatures. The rapid formation of orthoester, was most pronounced around −70 °C. Over time, rearrangement of the orthoester to the desired product is also evident.
 | ||
| Fig. 2 (a) Normalized HPLC results for glycosylation of nucleophile 1 with mannosyl donor 2. (b) Normalized HPLC results for glycosylation of nucleophile 5 with mannosyl donor 1. Legend, reaction times: ◊ 213.5 s, × 106.8 s,△53.4 s,○ 26.7 s. | ||
Following the success with the initial glycosylation, 2,3,4-tri-O-benzyl-methyl mannoside 5 was mannosylated with 2 (Scheme 1b). The more sterically hindered nucleophile is more difficult to glycosylate and contains a benzyl group that facilitates monitoring during HPLC analysis.
In contrast to the results obtained for the coupling of 1 and 2, microreactor-HPLC analysis of the union of 2 and 5 shows a unique reaction profile (Fig. 2b). Optimal product yields are obtained from −60 to −40 °C, the same temperature range that fosters orthoester formation. The reaction outcome is optimal at −60 °C with a reaction time of just over 213 seconds. However, this analysis also demonstrates that nearly the same yield is achievable by running the reaction at −35 °C for 25.7 seconds. With very little change in overall yield, it would be possible to increase production by nearly an order of magnitude over the slower reactions run at lower temperatures. The microreactor-HPLC study reveals important information regarding process development for scale-up, in addition to reaction optimization. From the perspective of developing a method for semi-preparative or preparative scale, significant advantage can be found from the results of this continuous-flow study, over a much more cumbersome and costly batchwise optimization.
Unlike batch methods, which are challenged by the difficulty of handling microliter quantities of volatile solvents and the possibility of external contamination, the enclosed microreactor system serves to rapidly obtain comprehensive information about a given transformation. With a single preparation of reagents, 44 reactions were completed at varying temperatures and reaction times requiring just over 2 mg of glycosylating agent for each reaction.
Notes and references
- J. Lehmann, in Carbohydrates: Structure and Biology, Thieme, Stuttgart, 1998, p. 1–45 Search PubMed.
- For review, see: K. Toshima and K. Tatsuta, Chem. Rev., 1993, 120, 1503 Search PubMed; T. Nukada, A. Berces, M. Z. Zgierski and D. M. Whitfield, J. Am. Chem. Soc., 1998, 120, 13291 CrossRef.
- R. Carlson, Design and Optimization in Organic Synthesis, Elsevier Science: Amsterdam, New York, 1992 Search PubMed.
- K. Jähnisch, V. Hessel, H. Löwe and M. Baerns, Angew. Chem. Int. Ed., 2004, 43, 406 CrossRef; H. Pennemann, P. Watts, S. J. Haswell, V. Hessel and H. Löwe, Org. Process Res. Dev., 2004, 8, 422 Search PubMed; P. D. I. Fletcher, S. J. Haswell, E. Pombo-Villar, B. H. Warrington, P. Watts, S. Y. F. Wong and X. Zhang, Tetrahedron, 2002, 58, 4735 CrossRef CAS.
- M. Schilling, W. Nigge, A. Rudzinski, A. Neyer and R. Hergenröder, Lab. Chip, 2004, 4, 220 RSC; R. J. Jackman, T. M. Floyd, R. Ghodssi, M. A. Schmidt and K. F. Jensen, J. Micromech. Microeng., 2001, 11, 263 CrossRef CAS; H. Lu, M. A. Schmidt and K. F. Jensen, Lab. Chip., 2001, 1, 22 RSC.
- K. F. Jensen, Chem. Eng. Sci., 2001, 56, 293 CrossRef CAS.
- M. W. Losey, R. J. Jackman, S. L. Firebaugh, M. A. Schmidt and K. F. Jensen, J. Microelectromech. Sys., 2002, 11, 709 Search PubMed.
- P. Fügedi, A. Lipták and P. N. Neszmélyi, Carbohydr. Res., 1982, 107, C5 CrossRef; A. Vasella, C. Witzig, C. Waldraff, P. Uhlmann, K. Briner, B. Bernet, L. Panza and R. Husi, Helv. Chim. Acta, 1993, 76, 2847 CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: experimental details. See http://www.rsc.org/suppdata/cc/b4/b414503h/ |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2005 |