A small peptide stereochemically customized as a globular fold with a molecular cleft†
Soumendra
Rana
a,
Bijoy
Kundu
b and
Susheel
Durani
*a
aDepartment of Chemistry, Indian Institute of Technology, Bombay, Mumbai-400076, India. E-mail: sdurani@iitb.ac.in
bMedicinal Chemistry Division, Central Drug Research Institute, Lucknow-226001, India
First published on 17th November 2004
Abstract
A boat shaped peptide molecular fold is generated by stereochemical modification of a 20-residue β-hairpin peptide, making it a promising prototype for future optimization as a molecular receptor.
The functional versatility of proteins stands in sharp contrast with their extremely conservative molecular architectural plans. The homochiral nature of polypeptide stereochemical structure restricts the choice of both residue and motif level conformations in proteins to a handful and therefore the possibilities for tertiary structure to ∼103 topologically distinct folds.1 We recently presented a stereochemical recipe for large scale diversification of peptide molecular architecture, which has a built-in prospect for customization of both molecular forms and functions. A bracelet shaped molecular fold was accomplished in a 14-residue peptide as an example of stereospecific design.2 Extending the concept, a boat shaped molecular fold is accomplished here in a 20-residue β-hairpin peptide, making it a smallest known globular fold with a molecular cleft, and a suitable template for future development as a molecular receptor. The β-hairpin is modelled with a type-II′ turn segment centered at D-Pro(10)–Gly(11), which stereochemically guides the antiparallel chain reversal3 for geometrically conducive inter-strand hydrogen bonding. Maximum number of β-sheet favoring residues are used in each strand known from statistical databases. Six long armed apolar residues are positioned at 1, 5, 9, 12, 16, and 20 so that stereochemical inversions from L-to-D chiral residues at positions 3, 18 and 7, 14 create upturned sections in both strands, resulting in a close juxtaposition of the six apolar side chains forming the hydrophobic core of a quasi-globular mini-protein with a largely hydrophilic exterior as illustrated in Fig. 1 and Fig. S1A, B and C†. Some side-by-side polar residues [Thr(8)–Ser(13), Asn(15)–Ser(6), and Lys(17)–Glu(4)] are so chosen as to enhance the structural and morphological stability of the molecular fold through inter-strand side-chain interactions, making it potentially water soluble with a hydrophobic type clustering in its central molecular cavity. The designed hairpin [Ac–Met(1)–Thr(2)–D-Val(3)–Glu(4)–Trp(5)–Ser(6)–D-Ala(7)–Thr(8)–Ile(9)–D-Pro(10)–Gly(11)–Val(12)–Ser(13)–D-Val(14)–Asn(15)–Leu(16)–Lys(17)–D-Ala(18)–Thr(19)–Leu(20)–NH2] (Fig. S1C†) responded well to a 2 ns unrestrained molecular dynamics (MD) simulation at 300K in explicit H2O with gromos-96 force field in the GROMACS software package,4 in contrast to “Trp-cage”, a recently designed mini-protein5 of comparable sequence length, suggesting the possibility of appreciable ordering cum morphological stability, perhaps one of the reasons being the comparatively superior hydrophobic driving force (ESI†). The peptide made by solid phase synthesis6 and identified by MALDI-MS, displayed the anticipated molecular morphology of a globular fold with a molecular cleft by the combined use of NMR, MD, fluorescence and CD.
 | ||
| Fig. 1 DYANA generated mean NMR model of the boat shaped molecular fold, displaying the clustering of six apolar side chains. | ||
NMR analysis7 in H2O established the presence of an inter-strand NOE signature, illustrated in Fig. 2, expected of a canonical β-hairpin peptide. Nearly 80% of all expected inter-strand long-range NOEs could be clearly identified proving the ordering of a β-hairpin like molecular fold in H2O. The expected NH–CαH or NH–CβH inter-strand NOEs that could be unambiguously assigned include those between Val(14)–Thr(8), Val(12)–Thr(8), Ala(7)–Asn(15), Leu(16)–Ser(6), Leu(16)–Glu(4), Ala(18)–Glu(4), Ala(18)–Thr(2), Leu(20)–Thr(2) and Met(1)–Thr(9). Further, a rich spectrum of NOEs involving the aromatic protons of Trp(5) and the side chains of the residues at positions 1, 2, 3, 9, 16, 18, 19 and 20 [Trp(5)HZ2–Met(1)CγH, Trp(5)HE3–Thr(2)CαH, Trp(5)HZ3γ–Thr(2)CαH, Trp(5)HH2–Thr(2)CβH, Trp(5)HZ2–Val(3)CαH, Trp(5)HD1–Ile(9)CβH, Trp(5)HE3–Leu(16)CβH, Trp(5)HZ2–Ala(18)CβH, Trp(5)HH2–Thr(19)CαH, Trp(5)HZ2–Thr(19)CαH, Trp(5)HZ3–Thr(19)CαH, Trp(5)HH2–Leu(20)NH and Trp(5)HZ3–Leu(20)NH] could be clearly observed, indicating the existence of a molecular cleft like conformation with a hydrophobic type clustering.
 | ||
| Fig. 2 Summary of long-range NOEs observed in H2O. | ||
Absence of NOEs between the side chains of Trp(5) and Val(12) and between the side chains of Met(1) and Leu(20), suggested the possibility of poorer hydrophobic clustering of these side chains. Torsion angle dynamics simulation was performed with DYANA8 using 35 selected distance restraints (intra 0, short 15, medium 5, long 15) calibrated on the basis of relative NOE intensities. Out of the 50 random structures gathered, MOLMOL9 superposed ten lowest energy structures are shown in Fig. S6†. There was an NOE violation of ≤0.58 Å in these structures and the mean global backbone root mean square deviation (RMSD) over residues 2–19 was a remarkable 0.39 ± 0.24 Å.
The energy minimized average DYANA structure of the peptide, shown in Fig. 1, was also submitted to a 16 ns unrestrained MD simulation at 300K in H2O. No significant changes occurred either in the radius of gyration of the peptide or in its potential energy over the MD trajectory, and a well-preserved network of hydrogen bonds was observed during this period, indicating the ordering of the peptide to an appreciable extent (ESI†).
The peptide displayed a fluorescence emission band in H2O due to tryptophan emission which is 4 nm blue shifted with respect to the external fluorophore probe NATA (N-acetyl-L-Tryptophan amide), as expected for a tryptophan held in a relatively non-polar environment.10 However, the peptide displayed an unusual two-fold lower intensity of tryptophan fluorescence as compared with the reference molecule NATA, which is anomalous for a tryptophan buried in a hydrophobic environment.11 The quenching of tryptophan fluorescence could be attributed to the effect of the sulfur atom in Met(1) side chain, which is in direct contact with the Trp(5) side chain with an average distance of 5.92 ± 0.09 Å from the centroid of the aromatic ring over the 10 best DYANA structures in comparison with the requirement of ≤7 Å for contact quenching of the tryptophan chromophore by sulfur atom.12 An independent experiment established that free methionine is an effective quencher of tryptophan fluorescence in H2O with a Stern–Volmer quenching constant (KSV) of 3.3 against NATA, in comparison with a KSV of 30 for KI, the standard quencher (ESI†). According to NMR and MD analysis only a boat shaped peptide molecular fold can explain this result since the side chains of Met(1) and Trp(5) are far away in a canonical planar β-sheet type structure with an average distance of 14.57 ± 2.55 Å between the centroid of aromatic ring in the Trp(5) side chain and the sulfur atom in the Met(1) side chain. Another evidence for hydrophobic clustering around the Trp(5) side chain in the peptide was obtained from its response to the effect of the external fluorescence quencher KI. A KSV of 19 for the peptide against KI, in comparison with a KSV of 30 in the case of NATA (ESI†) provided evidence that the Trp(5) side chain is partially buried in the folded peptide.13 The percentage of tryptophan burial assessed from this data is about 63%, while the burial calculated by NACCESS14 in an energy minimized mean NMR model with reference to a Gly–Trp–Gly model was 73%, and judged by MD in H2O over the 16 ns trajectory was also 73% (ESI†). Only 11% of this burial is contributed by the contact between Trp(5) and Met(1) side chains as judged by NACCESS analysis on mean NMR model generated by DYANA and based on MD analysis in the absence of Met(1).
The peptide displays a CD signature diagnostic of β-sheet structure15 with a negative band between 215–235 nm (Fig. 3). This pattern is consistent with the presence of a central β-sheet segment of canonical nature in this peptide, which was absent in our earlier reported 14-mer bracelet shaped hairpin,2 which also lacked the presence of a standard β-sheet type CD signature. The nature of the CD band remains more or less unchanged in both 70% trifluoroethanol–H2O and 70% methanol–H2O except for a 5 nm blue shift in presence of TFE (Fig. 3). These solvents are known for their strong conformation inducing effects in small fluxional peptides16 hence our boat shaped molecular fold could be experiencing a comparable level of ordering under all three solvent conditions, and presumably it is free from fluxional character in water.
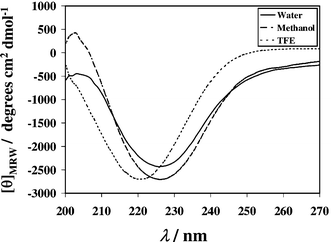 | ||
| Fig. 3 Solvent dependent far UV CD spectra of the peptide at 298K. | ||
A handful of ≤30 residue linear peptides reported in the recent literature, basically as sequence variants of known molecular folds of native like morphology, have evoked tremendous scientific interest, because of their mini-protein like globularity and two-state behavior in H2O.5,17 Implementing a simple and predictable departure from Nature's design recipe, a globular molecular fold of comparable size but radically different molecular morphology is achieved here, making it a more promising platform for functional design involving molecular recognition. With a central molecular cavity harboring six hydrophobic amino acid side chains, the further elaboration of receptor like functions could be a matter of sequence optimization in the presence of a desired guest molecule, for which powerful inverse sequence design procedures have recently been developed.18
This work was supported by a research grant from the Council of Scientific and Industrial Research, New Delhi, India. The use of NMR at National High Field NMR Facility, TIFR, Mumbai is highly acknowledged.
Notes and references
- (a) C. Chothia, Nature, 1992, 357, 543 CrossRef CAS; (b) Z. X. Wang, Protein Eng., 1998, 11, 621 CrossRef CAS.
- S. Rana, B. Kundu and S. Durani, Chem. Commun., 2004, 2462 RSC.
- (a) B. L. Sibanda and J. M. Thornton, Nature, 1985, 316, 170 CrossRef CAS; (b) H. E. Stanger and S. H. Gellman, J. Am. Chem. Soc., 1998, 120, 4236 CrossRef CAS.
- E. Lindahl, B. Hess and D. van der Spoel, J. Mol. Model., 2001, 7, 306 Search PubMed.
- J. W. Neidigh, R. W. Fesinmeyer and N. H. Andersen, Nat. Struct. Biol., 2002, 6, 425 CrossRef CAS.
- W. C. Chan and P. D. White, Fmoc Solid Phase Peptide Synthesis: A Practical Approach, IRL Press, Oxford, UK, 1989 Search PubMed.
- (a) D. G. Davis and A. Bax, J. Am. Chem. Soc., 1985, 107, 2821 CrossRef CAS; (b) A. Kumar, R. R. Ernst and K. Wuthrich, Biochem. Biophys. Res. Commun., 1980, 95, 1 CAS; (c) K. Wuthrich, NMR of Proteins and Nucleic Acids, Wiley, New York, 1986 Search PubMed.
- P. Guntert, C. Mumenthaler and K. Wuthrich, J. Mol. Biol., 1997, 273, 283 CrossRef CAS.
- R. Koradi, M. Billeter and K. Wuthrich, J. Mol. Graphics, 1996, 14, 51 CrossRef CAS.
- (a) J. R. Lakowicz, Principles of Fluorescence Spectroscopy, Plenum Press, New York, 1983 Search PubMed; (b) J. Y. Pan, J. C Sanford and M. W. Resnick, J. Biol. Chem., 1995, 41, 24204.
- V. Nanda, S. M. Liang and L. Brand, Biochem. Biophys. Res. Commun., 2000, 279, 770 CrossRef CAS.
- (a) T. Yuan, A. M. Weljie and H. J. Vogel, Biochemistry., 1998, 37, 3187 CrossRef CAS; (b) E. A. Meyer, R. K. Castellano and F. Diederich, Angew. Chem., Int. Ed., 2003, 42, 1210 CrossRef CAS.
- E. Rust, D. L. Martin and C. H. Chen, Arch. Biochem. Biophys., 2001, 392, 333 CrossRef CAS.
- S. J. Hubbard and J. M. Thornton, NACCESS, Computer Program, Department of Biochemistry and Molecular Biology, University College London, 1993 Search PubMed.
- G. D. Fasman, Circular Dichroism and the Conformational Analysis of Biomolecules, Plenum Press, New York, 1996 Search PubMed.
- (a) D. P. Hong, M. Hoshino, R. Kuboi and Y. Goto, J. Am. Chem. Soc., 1999, 121, 8427 CrossRef CAS; (b) B. Ciani, M. Jourdan and M. S. Searle, J. Am. Chem. Soc., 2003, 125, 9038 CrossRef CAS.
- (a) M. D. Struthers, R. P. Cheng and B. Imperiali, Science, 1996, 271, 342 CrossRef CAS; (b) B. I. Dahiyat and S. L. Mayo, Science, 1997, 278, 82 CrossRef CAS; (c) A. G. Cochran, N. J. Skelton and M. A. Starovasnik, Proc. Natl. Acad. Sci., 2001, 98, 5578 CrossRef CAS.
- (a) L. L. Looger, M. A. Dwyer, J. J. Smith and H. W. Helinga, Nature, 2003, 423, 185 CrossRef CAS; (b) D. N. Bolon and S. L. Mayo, Proc. Natl. Acad. Sci., 2001, 98, 14274 CrossRef CAS; (c) J. S. Marvin and H. W. Hellinga, Proc. Natl. Acad. Sci., 2001, 98, 4955 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: all experimental procedures, MALDI-MS, NMR, molecular dynamics, fluorescence and CD data of the peptide in H2O. See http://www.rsc.org/suppdata/cc/b4/b413802c/ |
| This journal is © The Royal Society of Chemistry 2005 |