Sequential homobimetallic catalysis: an unprecedented tandem Pd(0)-catalysed deprotection – Pd(II)-catalysed heterocyclisation reaction leading to benzofurans
Bartolo
Gabriele
*a,
Raffaella
Mancuso
b,
Giuseppe
Salerno
b and
Lucia
Veltri
b
aDipartimento di Scienze Farmaceutiche, Università della Calabria, 87036, Arcavacata di Rende (CS), Italy. E-mail: b.gabriele@unical.it; Fax: 39 0984 492044; Tel: 39 0984 492813
bDipartimento di Chimica, Università della Calabria, 87036, Arcavacata di Rende (CS), Italy
First published on 12th November 2004
Abstract
We report here the first example of “sequential homobimetallic catalysis”: a transition metal catalyst with the metal in a certain oxidation state catalyses the deprotection of a functional group, which in situ undergoes a subsequent transformation catalysed by another complex of the same metal but in a different oxidation state.
We report here the first example of “sequential homobimetallic catalysis”: a transition metal catalyst with the metal in a certain oxidation state catalyses the deprotection of a functional group, which in situ undergoes a subsequent transformation catalysed by another complex of the same metal but in a different oxidation state. The concept is illustrated by the tandem Pd(0)-catalysed carbonylative deallylation – Pd(II)-catalysed carbonylative cyclisation of 1-(2-allyloxyphenyl)-2-yn-1-ols 1, leading to 2-benzofuran-2-ylacetic methyl esters 2 in high yields together with but-3-enoic acid methyl ester 3 as coproduct, according to Eqn. (1).1
 | (1) |
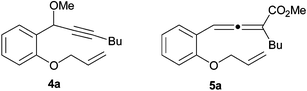
The first experiments were carried out with 1-(2-allyloxyphenyl)hept-2-yn-1-ol 1a (R1 = H, R2 = Bu) at 100 °C and under 30 atm of CO, in anhydrous MeOH as the solvent and nucleophile, and in the presence of Pd(PPh3)4 (0.5 mol %) and PdI2 (0.5 mol %) in conjunction with an excess of KI (100 equiv with respect to PdI2)2 as the catalytic systems. After 15 h, benzofuran-2-ylhexanoic acid methyl ester 2a was obtained as the main reaction product (76% GLC yield), together with 3 (ca. 70%) and small amounts of 1-allyloxy-2-(1-methoxyhept-2-ynyl)benzene 4a (6%, deriving from etherification of the alcoholic function of 1a) and of 4-(2-allyloxyphenyl)-2-butylbuta-2,3-dienoic acid methyl ester 5a (8%, from Tsuji-type carbonylation3 of the propargylic function of 1a).
The two sequential catalytic cycles leading to 2 and 3 are shown in Scheme 1 (unreactive ligands are omitted for clarity). The first cycle, catalysed by Pd(0), corresponds to the oxidative addition of the allyloxy moiety to Pd(0) followed by cleavage by iodide anions and allylic carbonylation,4 with formation of 3 and 2-(1-hydroxyalk-2-ynyl)phenate. The latter then acts as substrate in the second catalytic cycle, catalysed by Pd(II): a 5-exo-dig type heterocyclisation occurs through nucleophilic attack by oxygen on the triple bond activated by coordination to Pd(II) followed by methoxycarbonylation,5 leading to intermediate I and H–Pd–I. Reduction of the allylic alcohol moiety of I then takes place by the reaction of I with H–Pd–I (with formation of a π-allyl complex and elimination of water),6 followed by regiospecific protonolysis7 with formation of 2 and regeneration of the Pd(II) catalyst.
 | ||
| Scheme 1 Sequential homobimetallic catalysis leading to 2. | ||
Several experimental evidences support the validity of the proposed catalytic sequence. (a) First of all, the allenic derivative 5a was not an intermediate in the formation of 2a, since it was not converted into 2a under the reaction conditions. This result rules out the possibility that carbonylation occurs first, followed by deallylative cyclisation. (b) Practically no deallylation occurred when the reaction was carried out under the same conditions reported above, but in the absence of Pd(PPh3)4: with 1 mol % of PdI2 along with 100 equiv of KI and 4 equiv of PPh3 in anhydrous MeOH, 2a was formed in only 8% GLC yield, the main reaction product being 4a (83% GLC yield; 5a was also present in the reaction mixture in 2% yield). This result confirms the essential role played by Pd(0) in promoting the initial deallylation step. (c) When the reaction was carried out with Pd(PPh3)4 in the absence of the PdI2–KI catalyst, only traces of 2a were obtained. This result shows that no carbonylative cyclisation occurs in the absence of the Pd(II) catalyst.
Interestingly, we have found that a PPh3-stabilized Pd(0) complex could be formed in situ directly from PdI2 and PPh3 working in MeOH in the presence of small amounts of H2O. In fact, under these conditions, formation of an I–Pd–CO2H species (from the reaction between PdI2, CO and H2O)8 occurs, whose decarboxylation9 affords H–Pd–I in equilibrium with Pd(0) and HI. Actually, the use of PdI2 (1 mol %) in conjunction with 100 equiv of KI, 4 equiv of PPh3 and 200 equiv of H2O (at 100 °C and under 30 atm of CO, as in the previous experiments) led to even better results with respect to the PdI2–KI–Pd(PPh3)4 system: after 15 h, 2a was obtained as the sole product in 96% GLC yield [91% isolated, Eqn. (1); this result should be compared with the 76% GLC yield obtained above with the PdI2–KI–Pd(PPh3)4 system]. The presence of PPh3 was essential for the reaction, its function being to stabilize the Pd(0) species responsible for the initial deallylation. In fact, by carrying out the above reaction without PPh3, the main reaction product was 4a (85% GLC yield), benzofuran 2a being formed in only 7% GLC yield along with small amounts of 5a (2%). Under the same conditions optimized for the reaction of 1a, other 1-(2-allyloxyphenyl)-2-yn-1-ols 1b–d were easily converted, after 15–24 h, into the corresponding benzofuran-2-ylacetic esters 2b–d in high isolated yields [80–82%, Eqn. (1)].† It is noteworthy that the reaction worked nicely even with a very bulky substituent on the triple bond, as in the case of 1c (R2 = t-Bu).
In conclusion, we have reported an unprecedented catalytic sequence involving two sequential catalytic cycles: in the first cycle, promoted by Pd(0), deprotection of a nucleophilic oxygen occurs, with formation of the substrate1 undergoing the subsequent carbonylative heterocyclisation process, catalysed by Pd(II). From a synthetic point of view, the net transformation corresponds to the one-step, selective conversion of simple and readily available starting materials10 into very important heterocyclic derivatives in high yields. Benzofurans are in fact a very important class of heterocycles, which display a wide range of biological activity.11 In particular, benzofuranacetic derivatives are known to exhibit a peculiar and very interesting pesticidal, insecticidal, and acaricidal activity.12
Notes and references
- 2-(1-Hydroxyalk-2-ynyl)phenols, with the –OH group unprotected, could not be prepared and used directly as substrates because of their instability, according to the literature: D. Pflieger and B. Muckensturm, Tetrahedron Lett., 1990, 31, 2299 Search PubMed.
- The use of PdI2 in conjunction with an excess of KI as the catalytic system for carbonylation reactions was disclosed by us several years ago: B. Gabriele, M. Costa, G. Salerno and G. P. Chiusoli, Chem. Commun., 1992, 1007 Search PubMed; B. Gabriele, M. Costa, G. Salerno and G. P. Chiusoli, J. Chem. Soc., Perkin Trans. 1, 1994, 83 RSC.
- J. Tsuji and T. Mandai, J. Organomet. Chem., 1993, 451, 15 CrossRef CAS and references therein.
- The Pd(0) catalysed substitutive carbonylation of allylic derivatives is a well-known reaction: J. Tsuji, Palladium Reagents and Catalysts, John Wiley & Sons, Chichester, 1995, pp. 340–345 Search PubMed; T. Mandia, in Handbook of Organopalladium Chemistry for Organic Synthesis, ed. E. Negishi, Wiley-InterScience, New York, 2002, vol. 2, pp. 2505–2508 Search PubMed.
- For very recent reviews and accounts on this kind of reactivity, see: B. Gabriele, G. Salerno, M. Costa and G. P. Chiusoli, J. Organomet. Chem., 2003, 687, 219 Search PubMed; B. Gabriele, G. Salerno, M. Costa and G. P. Chiusoli, Curr. Org. Chem., 2004, 8, 919 CrossRef CAS; F. Alonso, I. Beletskaya and M. Yus, Chem. Rev., 2004, 104, 3079 CAS; S. A. Vizer, K. B. Yerzhanov, A. A. A. Al Quntar and V. M. Vembitsky, Tetrahedron, 2004, 60, 5499 CrossRef CAS.
- The possibility of obtaining a π-allylpalladium complex directly from the reaction between an allyl alcohol and a palladium hydride species without any activator was disclosed by us some years ago: B. Gabriele, G. Salerno, M. Costa and G. P. Chiusoli, J. Mol. Catal., 1996, 111, 43 Search PubMed . More recently, the formation of an allylpalladium intermediate from allyl alcohols and an hydridopalladium complex has been reported: F. Ozawa, T. Ishiyama, S. Yamamoto, S. Kawagishi and H. Murakami, Organometallics, 2004, 23, 1698 Search PubMed.
- The possibility of reducing an allyl alcohol moiety through the reaction with an H–Pd–I species with formation of a π-allyl complex followed by protonolysis has been recently demonstrated by us: G. P. Chiusoli, M. Costa, L. Cucchia, B. Gabriele, G. Salerno and L. Veltri, J. Mol. Catal. A: Chem., 2003, 687, 219 Search PubMed.
- B. Gabriele, L. Veltri, G. Salerno, M. Costa and G. P. Chiusoli, Eur. J. Org. Chem., 2003, 1722 CrossRef CAS and references therein.
- Decarboxylation of X–Pd–CO2H complexes to give X–Pd–H species is a well-known process: see, for example, references 6, 7 and R. Bertani, G. Cavinato, L. Toniolo and G. Vasapollo, J. Mol. Catal., 1993, 84, 165 Search PubMed; V. V. Grushin, Chem. Rev., 1996, 96, 2011 CrossRef CAS.
- Substrates 1 were easily prepared in 86–92% overall isolated yields starting from commercially available 2-hydroxybenzaldehyde (R1 = H) or 2′-hydroxyacetophenone (R1 = Me) through allylation (as described by B. S. Orlek, P. G. Sammes and D. J. Weller, Tetrahedron, 1993, 49, 8179 Search PubMed ) followed by Grignard reaction with R2C
![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) CMgBr.
CMgBr. - For some very recent examples, see: E. Tsuji, K. Ando, J. Kunitomo, M. Yamashita, S. Ohta, S. Kohno and Y. Ohishi, Org. Biomol. Chem., 2003, 1, 3139 Search PubMed; K. Kawasaki, M. Masubuchi, K. Morikami, S. Sogabe, T. Aoyama, H. Ebiike, S. Niizuma, M. Hayase, T. Fujii, K. Sakata, H. Shindoh, Y. Shiratori, Y. Aoki, T. Ohtsuka and N. Shimma, Bioorg. Med. Chem. Lett., 2003, 13, 87 RSC.
- T. N. Wheeler (Union Carbide Corp., USA), US Pat. 4,431,650, 1977 (Chem. Abstr. 1984, 101, 54903u) Search PubMed.
Footnote |
| † Representative experimental procedure for the synthesis of 2: a 250 mL stainless steel autoclave was charged with PdI2 (5.0 mg, 1.39·10−2 mmol), KI (230 mg, 1.39 mmol), PPh3 (14.6 mg, 5.57·10−2 mmol) and a solution of 1 (1.40 mmol) in anhydrous MeOH (6.3 mL). Water (50 µL, 2.78 mmol) was then added, and the autoclave was sealed, purged at room temperature several times with CO with stirring (5 atm) and eventually pressurized at 30 atm. After stirring at 100 °C for 15 h (1a–c) or 24 h (1d), the autoclave was cooled and degassed. The solvent was evaporated and products were purified by column chromatography [SiO2, 1 ∶ 1 hexane–CH2Cl2 (2a), 8 ∶ 2 hexane–acetone (2b), 8 ∶ 2 hexane–AcOEt (2c), 9 ∶ 1 hexane–AcOEt (2d)]. Characterization data for 2a (315 mg, 91%, colourless oil): IR (film): ν = 1743, 1454, 1252, 1160, 751 cm−1; 1H NMR (300 MHz, CDCl3): δ = 7.53–7.49 (m, 1 H), 7.47–7.42 (m, 1 H), 7.27–7.15 (m, 2 H), 6.59–6.58 (m, 1 H), 3.82 (t, J = 7.3 Hz, 1 H), 3.72 (s, 3 H), 2.18–1.93 (m, 2 H), 1.43–1.24 (m, 4 H), 0.89 (t, J = 6.8 Hz, 3 H); 13C NMR (75 MHz, CDCl3): δ = 172.1, 155.3, 154.8, 128.4, 123.9, 122.7, 120.7, 111.1, 103.8, 52.3, 45.7, 30.6, 29.5, 22.4, 13.8; MS (EI, 70 eV): m/z (%): 246 (33) [M+], 187 (36), 131 (100). For 2b (303 mg, 81%, yellow oil): IR (film): ν = 1739, 1453 1253, 1156, 751 cm−1; 1H NMR (300 MHz, CDCl3): δ = 7.50–7.26 (m, 7 H), 7.25–7.12 (m, 2 H), 6.57 (t, J = 1.0 Hz, 1 H), 5.14 (s, br, 1 H), 3.74 (s, 3 H); 13C NMR (75 MHz, CDCl3): δ = 170.5, 155.0, 154.6, 128.8, 128.7, 128.2, 128.0, 124.1, 123.6, 122.7, 120.9, 111.1, 105.2, 52.6, 51.7; MS (EI, 70 eV): m/z (%) 266 (20) [M+], 207 (100), 178 (31). For 2c (275 mg, 80%, pale yellow solid, mp 60–61 °C): IR (KBr): ν = 1733, 1456, 1243, 1205, 1150, 755, 747 cm−1; 1H NMR (300 MHz, CDCl3): δ = 7.54–7.49 (m, 1 H), 7.46–7.41 (m, 1 H), 7.25–7.14 (m, 2 H), 6.74 (dd, J = 1.0 Hz, 0.3 Hz, 1 H), 3.73 (d, J = 0.3 Hz, 1 H), 3.69 (s, 3 H), 1.08 (s, 9 H); 13C NMR (75 MHz, CDCl3): δ = 171.2, 154.5, 153.9, 128.5, 123.7, 122.7, 120.7, 111.0, 105.5, 55.9, 51.7, 35.1, 28.0. MS (EI, 70 eV): m/z (%): 246 (11) [M+], 190 (100), 158 (35). For 2d (298 mg, 82%, pale yellow oil): IR (film): ν = 1743, 1455, 1255, 1246, 1167, 747 cm−1; 1H NMR (300 MHz, CDCl3): δ = 7.47–7.40 (m, 2 H), 7.27–7.17 (m, 2 H), 3.84 (dd, J = 9.1 Hz, 6.6 Hz, 1 H), 3.68 (s, 3 H), 2.21 (s, 3 H), 2.23–1.96 (m, 2 H), 1.41–1.17 (m, 4 H), 0.87 (t, J = 7.1 Hz, 3 H); 13C NMR (75 MHz, CDCl3): δ = 172.1, 154.1, 149.6, 130.0, 123.8, 122.2, 119.0, 112.2, 111.1, 52.2, 43.6, 29.7, 29.5, 22.4, 13.9, 7.9; MS (EI, 70 eV): m/z (%) 260 (23) [M+], 201 (53), 145 (100). Elemental analyses were satisfactory. |
| This journal is © The Royal Society of Chemistry 2005 |