Synthesis of cyclopropanes via organoiron methodology: preparation of the C9–C16 alkenylcyclopropane segment of ambruticin†
Julie M.
Lukesh
and
William A.
Donaldson
*
Department of Chemistry, Marquette University, P.O. Box 1881, Milwaukee, WI 53201-1881, USA. E-mail: william.donaldson@marquette.edu; Fax: 414-288-7066; Tel: 414-288-7374
First published on 23rd November 2004
Abstract
A synthesis of the C9–C16 segment of ambruticin is described which relies on organoiron methodology to establish the 1,2,3-trisubstituted cyclopropane ring.
A variety of natural products and pharmaceuticals contain a substituted cyclopropane ring, and numerous synthetic routes to this functionality have been developed.1 We have recently reported on the scope and mechanism of a novel, iron mediated methodology for the preparation of 1,2,3-trisubstituted cyclopropanes (Scheme 1).2 This methodology relies on nucleophilic addition of stabilized carbon nucleophiles to (1-methoxycarbonylpentadienyl)iron cation 1 to generate (pentenediyl)iron complexes 2. The oxidative induced-reductive elimination of complexes 2 affords vinylcyclopropane carboxylates 3. Herein we report on the reaction of cations 1 with methyl nucleophiles and the subsequent oxidative decomplexation. The resultant cyclopropane product was utilized in synthesis of the C9–C16 alkenylcyclopropane segment of ambruticin 4, an orally active antifungal agent isolated from Polyangium cellulosum var. fulvum.3
 | ||
| Scheme 1 Synthesis of vinylcyclopropanes via organoiron methodology. | ||
Reaction of the tricarbonyl ligated cation 1a with dimethylcuprate gave diene complex 6a along with a minor amount of (pentenediyl)iron complex 5a (Table 1). In contrast, reaction of 1a with CH3Li in CH2Cl2 gave predominantly the (pentenediyl)iron complex 5a along with variable amounts of the known4 (methyl 3,5-hexadienoate)Fe(CO)3 (7a), while reaction of the dicarbonyl(triphenylphosphine) ligated cation 1b with MeLi/CH2Cl2 gave the pentenediyl complex 5b. The structures of pentenediyl complexes 5a/b and diene complex6a were assigned on the basis of their NMR spectral data. In particular, for the pentenediyl complexes 5a/b, the methyl resonance for each (δ 0.70 and 0.61 ppm respectively) appears as a doublet, indicative of only a single adjacent non-equivalent proton. Additionally, a 13C NMR signal at ca.δ 13–15 ppm and a 1H NMR signal at ca.δ 0.0 (d) ppm are characteristic of a carbon σ-bonded to iron and its attached proton.5 For the diene complexes 6a, the signal for the methyl protons (δ 0.96 ppm) appears as a triplet, indicative of two adjacent non-equivalent protons. Additionally, two 1H NMR at δ 6.05 (dd) and 5.26 (dd) ppm and two 13C NMR signals at δ 92.5, 85.5 ppm, are characteristic of an (η4-E-Z-dienoate)iron complex.5
 | (1) |
Formation of the products is rationalized by initial single electron-transfer from either methylcuprate or methyl lithium to afford a (pentadienyl)iron radical 8 and methyl–metal radical 9 (Scheme 2). Kochi has previously reported that certain nucleophilic additions to (pentadienyl)iron cations proceed via initial electron-transfer.6 In the case of methylcuprate collapse of the radical pair occurs via C–C bond formation at the terminal carbon, while for methyl lithium collapse of the radical pair occurs via C–C bond formation at the internal C2 carbon. If the radical pair 8 ∶ 9 escapes the solvent cage, then a second single electron transfer to 8 generates the pentadienyl anion 10. Aqueous work-up of the reaction mixture gives the protonated product 7. Notably, we have previously demonstrated the generation and alkylation of the (pentadienyl)iron anion 10 by deprotonation of 7.4
 | ||
| Scheme 2 Mechanism for addition of methyl nucleophiles. | ||
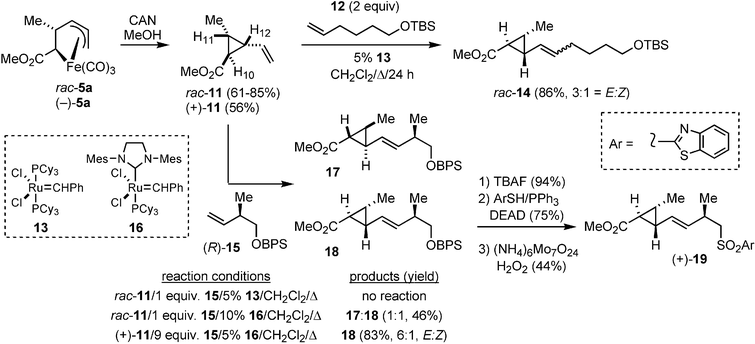
Oxidatively induced-reductive elimination of 5a with excess ceric ammonium nitrate (CAN) cleanly gave the vinylcyclopropane 11 (Scheme 3). The relative stereochemistry of 11 was assigned on the basis of its 1H NMR coupling data. The large coupling (ca. 9.6 Hz) between H11 and H12 (ambruticin numbering) indicates a cis relationship while smaller couplings between H10 and H11 and between H10 and H12 (ca. 4.9 Hz each) indicate a trans relationship.7 Preparation of optically active (+)-11 was accomplished in a similar fashion from the optically active cation (1S)-2.8
 | ||
| Scheme 3 Oxidatively induced-reductive elimination and olefin cross-metathesis. | ||
Introduction of the C13–C14 linkage by olefin cross-metathesis8,10 was envisioned. Reaction of rac-11 with 12
(2 equiv.) in the presence of (PCy3)2Cl2Ru![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CHPh (13, 10 mol%) gave alkenylcyclopropane 14
(86%) as a mixture of E- and Z-isomers (Scheme 3). The isolation of greater than a statistical yield of the cross-metathesis product indicates that the vinylcyclopropane 11 may be considered a “type-II” olefin in terms of its reactivity.9 In comparison, reaction of rac-11 with (R)-15
(1 equiv.)11 in the presence of 13
(5 mol%) gave no metathesis product after 24 h at reflux. Use of the more active IMes(PCy3)Cl2RuCHPh (16, 10 mol%) gave an inseparable mixture of diastereomeric alkenylcyclopropanes 17 and 18
(46%), along with homodimers resulting from self-metathesis (ca. 45% combined yield of homodimers). This statistical ratio of products indicates that 11 and 15 have comparable rates of cross-metathesis and homodimerization. With these results in hand, cross-metathesis of (+)-11 with a nine-fold excess of (R)-15 gave only 18 as a mixture of E- and Z-isomers (6 ∶ 1 ratio, 83% yield). Transformation of 18 into the sulfone 19 was accomplished by cleavage of the silyl ether, Mitsunobu reaction of the primary alcohol with 2-mercaptobenzothiazole, and finally oxidation with ammonium molybdate tetrahydrate.
CHPh (13, 10 mol%) gave alkenylcyclopropane 14
(86%) as a mixture of E- and Z-isomers (Scheme 3). The isolation of greater than a statistical yield of the cross-metathesis product indicates that the vinylcyclopropane 11 may be considered a “type-II” olefin in terms of its reactivity.9 In comparison, reaction of rac-11 with (R)-15
(1 equiv.)11 in the presence of 13
(5 mol%) gave no metathesis product after 24 h at reflux. Use of the more active IMes(PCy3)Cl2RuCHPh (16, 10 mol%) gave an inseparable mixture of diastereomeric alkenylcyclopropanes 17 and 18
(46%), along with homodimers resulting from self-metathesis (ca. 45% combined yield of homodimers). This statistical ratio of products indicates that 11 and 15 have comparable rates of cross-metathesis and homodimerization. With these results in hand, cross-metathesis of (+)-11 with a nine-fold excess of (R)-15 gave only 18 as a mixture of E- and Z-isomers (6 ∶ 1 ratio, 83% yield). Transformation of 18 into the sulfone 19 was accomplished by cleavage of the silyl ether, Mitsunobu reaction of the primary alcohol with 2-mercaptobenzothiazole, and finally oxidation with ammonium molybdate tetrahydrate.
In summary, a short route to the C9–C16 alkenylcyclopropane segment (19) of the structurally complex antifungal agent ambruticin was developed based on organoiron methodology.
Financial support from the National Science Foundation (CHE-0415771) and the Department of Education (P200A000228) is gratefully acknowledged. High resolution mass spectral determinations were made at the Washington University Mass Spectrometry Resource, an NIH Research Resource (Grant P41RR0954). The authors thank Dr Young K. Yun for preliminary experiments.
Notes and references
- For reviews see: J. Salaun, Top. Curr. Chem., 2000, 207, 1 Search PubMed; C. Cativiela and D. Diaz-de-Villegas, Tetrahedron: Asymmetry, 2000, 11, 645 CAS; R. E. Taylor, F. C. Engelhardt and M. J. Schmitt, Tetrahedron, 2003, 59, 5623 CrossRef CAS; H. Lebel, J.-F. Marcoux, C. Molinaro and A. B. Charette, Chem. Rev., 2003, 103, 977 CrossRef CAS.
- Y. K. Yun, K. Godula, Y. Cao and W. A. Donaldson, J. Org. Chem., 2003, 68, 901 CrossRef CAS.
- Isolation and structural determination: D. T. Connor, R. C. Greenough and M. von Strandtmann, J. Org. Chem., 1977, 42, 3664 Search PubMed; G. Just and P. Potvin, Can. J. Chem., 1980, 58, 2173 CrossRef CAS . Total syntheses: A. S. Kende, J. S. Mendoza and Y. Fujii, J. Am. Chem. Soc., 1990, 112, 9645 CAS; T. A. Kirkland, J. Colucci, L. S. Geraci, M. A. Marx, M. Schneider, D. E. Kaelin, Jr. and S. F. Martin, J. Am. Chem. Soc., 2001, 123, 12432 Search PubMed; E. Lee, S. J. Choi, H. Kim, H. O. Han, Y. K. Kim, S. J. Min, S. H. Son, S. M. Lim and W. S. Jang, Angew. Chem., Int. Ed., 2002, 41, 176 CrossRef CAS; P. Liu and E. N. Jacobsen, J. Am. Chem. Soc., 2001, 123, 10772 CrossRef CAS . For a compilation of references on synthetic studies see: V. Michelet, K. Adiey, S. Tanier, G. Dujardinand and J.-P. Genet, Eur. J. Org. Chem., 2003, 2947 CrossRef CAS.
- J. T. Wasicak, R. A. Craig, R. Henry, B. Dasgupta, H. Li and W. A. Donaldson, Tetrahedron, 1997, 53, 4185 CrossRef CAS.
- W. A. Donaldson, L. Shang, C. Tao, Y. K. Yun, M. Ramaswamy and V. G. Young, Jr., J. Organomet. Chem., 1997, 539, 87 CrossRef CAS.
- R. E. Lehmann, T. M. Bockman and J. K. Kochi, J. Am. Chem. Soc., 1990, 112, 458 CrossRef CAS; R. E. Lehmann and J. K. Kochi, Organometallics, 1991, 10, 190 CrossRef CAS.
- R. M. Silverstein, G. C. Bassler and T. C. Morrill, Spectrometric Identification of Organic Compounds, John Wiley & Sons, Inc., New York, NY, 1991 Search PubMed.
- (1S)-1a was prepared from (+)-tricarbonyl(methyl 6-oxo-2,4-hexadienoate)iron. C. Tao and W. A. Donaldson, J. Org. Chem., 1993, 58, 2134 Search PubMed; K. Godula, H. Bärmann and W. A. Donaldson, J. Org. Chem., 2001, 66, 3590 CrossRef CAS.
- A. K. Chatterjee, T.-L. Choi, D. P. Sanders and R. H. Grubbs, J. Am. Chem. Soc., 2003, 125, 10103 CrossRef CAS.
- For other examples of olefin cross-metathesis of vinylcyclopropanes see: C. Verbicky and C. K. Zercher, Tetrahedron Lett., 2000, 41, 8723 Search PubMed; T. Itoh, K. Mitsukuru, N. Ishida and K. Uneyama, Org. Lett., 2000, 2, 1431 CrossRef CAS.
- K. Konno, T. Fujishima, S. Maki, Z. Liu, D. Miura, M. Chokki, S. Ishizuka, K. Yamaguchi, K. Yan, M. Kurihara, N. Miyata, C. Smith, H. F. DeLuca and H. Takayama, J. Med. Chem., 2000, 43, 4247 CrossRef CAS.
Footnote |
| † This manuscript is dedicated to Prof. Michael A. McKinney on the occasion of his 65th birthday. |
| This journal is © The Royal Society of Chemistry 2005 |