DOI:
10.1039/B408479A
(Feature Article)
Chem. Commun., 2005, 23-36
Conducting metallopolymers: the roles of molecular architecture and redox matching
Received
(in Cambridge, UK)
4th June 2004
, Accepted 17th August 2004
First published on 18th October 2004
Abstract
Recent reports of highly conductive metallopolymers are reviewed. This literature is classified into one of two categories (inner or outer sphere) depending on the mode of interaction between the transition metal centers with each other and the conducting polymer backbone. The critical nature of charge transport is discussed in the context of the relative energies of the organic polymer-based and metal-centered redox processes. Also included are recent advances in the development of functional materials based on metal-containing conducting polymers.
| Bradley J. Holliday was born in West Lafayette, Indiana in 1975. He earned his BS in Chemistry from Allegheny College in Meadville, Pennsylvania in 1997. He completed his PhD at Northwestern University in 2002 under the supervision of Prof. Chad A. Mirkin in the area of supramolecular coordination chemistry. He is currently a postdoctoral research associate in the group of Prof. Timothy Swager. His research interests are in the areas of functional supramolecular and polymeric materials. |
| Timothy M. Swager is a Professor of Chemistry and the Associate Director of the Institute for Soldier Nanotechnologies at the Massachusetts Institute of Technology. A native of Montana, he received a BS from Montana State University in 1983 and a PhD from the California Institute of Technology in 1988. After a postdoctoral appointment at MIT, he was on the chemistry faculty at the University of Pennsylvania as an Assistant Professor 1990–1996 and as a Professor 1996. He moved to MIT in July of 1996 as a Professor of Chemistry. Swager's research interests are in design, synthesis, and study of organic-based electronic, sensory, and liquid crystalline materials. In the field of liquid crystals he developed new designs based upon shape complementarity to generate specific interactions and order between molecules. Swager's research in electronic polymers has been directed at the demonstration of new conceptual approaches to the construction of sensory materials and actuators. |
Introduction: architectures of metal-containing conducting polymers
Metal-containing conducting polymers
The study of conducting polymers has blossomed into a mature field over the last six decades.1 Tremendous progress has been made towards the goal of developing functional organic materials with delocalized π-electrons serving as the means of electronic conductivity.2 In such systems, chemical or electrochemical doping produces charged species within the polymer backbone and the mobility of these charges defines the bulk conductivity of a given material. In organic based materials, charge is efficiently shuttled due to isoenergetic states throughout the polymer backbone that can be interrupted by defects in the polymer strand (i.e., non-conjugated linkages, short conjugation length, or chemically altered monomer units). The vast majority of investigations performed in this field to date have dealt with purely organic frameworks and electron/hole transport in these systems is reasonably well understood. The incorporation of transition metals has the potential to greatly expand the function and ultimate applications of conducting polymer systems. More specifically, there is a steady and growing effort to incorporate redox-active metal centers into conducting polymer structures to create highly efficient redox conductivity for sensory (i.e., anions and small molecules), catalytic, photochemical, and photoelectronic applications. In conducting metallopolymers an understanding of the interactive roles that the metal centers and the organic polymer backbone play is in its early stages. Metal centers can provide efficient sites for redox conductivity, but can also provide thermodynamic sinks that trap/localize charges due to the introduction of low-lying energetic states. A few select systems have demonstrated important applications, however the potential of conducting metallopolymers is largely unrealized. To encourage progress towards realizing the full potential of these hybrid metal–organic conductors, we summarize herein recent work and highlight the general principles that need to be met in order to generate highly conductive metallopolymers.
Mechanisms of conductivity in metallopolymers
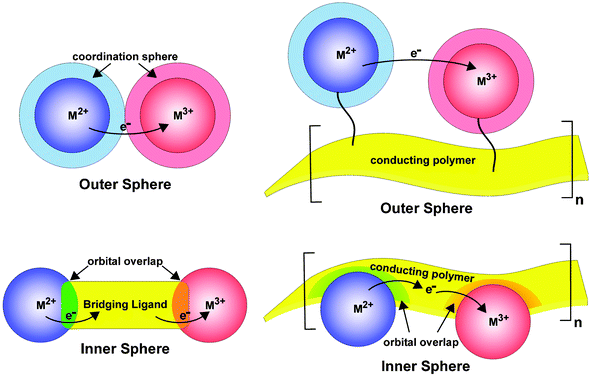
For the purposes of discussion, we emphasize two distinctly different redox conductivity mechanisms that pervade the conducting metallopolymer literature. In accord with classic inorganic electron transfer theory,3,4Fig. 1A details outer and inner sphere electron transfer mechanisms in mixed valence systems (top and bottom, respectively). The outer sphere mechanism is distinguished by the lack of mixing of the respective metal orbitals. In contrast, the inner sphere mechanism involves the communication of the two metal centers by orbital overlap via a mutually bridging ligand. It is important to note that the rate of electron transfer by this mechanism is highly dependent on the nature of the bridging ligand and its orbital overlap with the two metal centers.3 In conducting polymer systems incorporating redox-active transition metal centers we also find two unique environments. In the outer sphere arrangement, there are redox-active metals centers or complexes that decorate the periphery of the conducting polymer strand but have no direct interaction with the delocalized orbitals of the conducting organic polymer backbone, Fig. 1B
(top). Although the transition metals in this system may not be intimately involved in the conjugative pathway, they can still provide important charge transfer mechanisms. Analogous to molecular systems, in outer sphere architectures the metal centers are often tethered to the conducting polymer and have independent coordination spheres. In contrast, inner sphere architectures involve the incorporation of the transition metal centers into the polymer backbone with strong coupling between the orbitals of the transition metal and those involved in charge transport through the polymer strand, Fig. 1B
(bottom). When the energies of the orbitals are equivalent (same redox potential or redox matched) strong coupling provides for efficient additional charge transport pathways that intimately involve the transition metal centers leading to highly conductive materials.
General examples
There exists a diverse collection of systems that incorporate transition metals into conducting polymer structures. Of the two arrangements outlined above, the outer sphere arrangement has been the more widely studied. Recently systems have been reported that tether complexes such as Ru(bpy)3
(bpy = bipyridine) or ferrocene to the polymer backbone as shown in Fig. 2. As shown, the length and nature of the tether has been varied as well as the nature of the conducting polymer backbone. In most cases, a saturated tether has been employed resulting in electronic isolation of the metal complexes from the polymer backbone. From a molecular design point of view, the use of “well-behaved” redox-active metal complexes is easily understood since their electrochemical behavior is predicable. The electrochemical properties of several of these architectures will be outlined below.
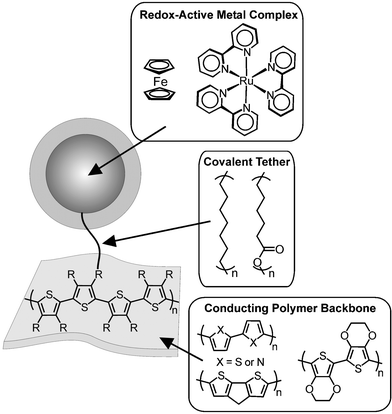 |
| | Fig. 2 Molecular components of outer sphere metallopolymer systems. | |
Although there are fewer examples of inner sphere type conducting metallopolymers, a rich structural diversity remains. Fig. 3 summarizes the general architectures of inner sphere systems with several specific examples from the recent literature superimposed. It is important to note that the transition metal center is directly interacting with the conjugation pathway, in some cases as an integral structural unit in the polymer framework. A variety of ligands can produce direct orbital overlap between the polymer backbone and the transition metal center thereby yielding materials with electronic properties that are highly sensitive to electronic perturbations at the metal center. The metal-containing functionalities can be varied widely and are not limited to those displaying metal-centered redox activity.
 |
| | Fig. 3 Molecular components of inner sphere metallopolymer systems. | |
Scope of this review
We provide herein a brief survey of recent conducting metallopolymer literature and the relative modes of conductivity. We will limit this review to materials with electrochemical properties/measurements that enable discussions of the degree of redox matching and its effect on conductivity. More comprehensive reviews of metal-containing polymeric materials and their model complexes have appeared elsewhere.5–15
Properties of metal-containing conducting polymers
Outer sphere electronic coupling
In the outer sphere arrangement the transition metal complexes are not directly involved in the conductivity pathway. However, in many cases the metal centers remain in electronic communication with the conducting polymer backbone through resonance or inductive effects. There are two prevalent synthetic strategies to accomplish an outer sphere arrangement. One involves non-conjugated tethers to attach the redox active metal centers to polymerizable monomer units. In the second strategy metal complexes are more intimately fused to the periphery of the monomer unit. Several examples of each will be discussed in the follow sections.
Tethered ferrocene systems
Over the past decade, a body of work has been developed concerning the functionalization of electropolymerizable groups ranging from parent thiophene and pyrrole to cyclopentabithiophene units with ferrocene.16–20 Several of these structures are summarized in Chart 1. Structures 1 and 3 require copolymerization with other thiophene-based monomers to form electroactive polymer films with ferrocene-based electrochemical responses.16,20 The study of the mixed monomer materials is inherently more difficult due to the poorly defined nature of the conducting polymer films, nevertheless separate electrochemical responses are observed for the polymer backbone and the pendent ferrocene. Zotti and coworkers have reported that a series of monomers, 2 and 4, electropolymerize cleanly to provide well-defined polymer films that have been studied by cyclic voltammetry, UV-Vis spectroelectrochemistry, and conductivity.17–19 The redox and conductivity properties of these materials are highly dependent on both the length and nature of the tethering moiety with conductivities in the range of 3 × 10−3
− 40 S cm−1. As the length of the alkyl tether is decreased the transport switches from a direct ferrocene-ferrocene self-exchange mechanism to one involving the conducting polymer backbone. Utilizing a conjugated alkene tether (not shown) further enhances the mechanism of conductivity that involves the polymer backbone directly. These studies provide a clear example of the outer sphere mechanism with the properties of the metal complexes and the conducting polymer backbone remaining electronically independent. The incidental overlap of the redox properties of the two components does not lead to a continuum of electronic states but instead an arbitrary cooperation wherein the metal complex and polymer backbone continue to function independently.
 |
| | Chart 1 | |
Tethered bipyridyl systems
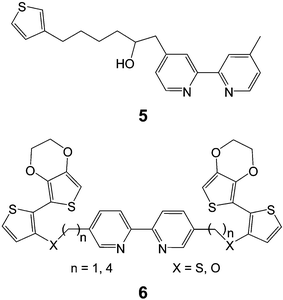
Ruthenium bipyridyl complexes are robust and electrochemically stable leading to their selection as attractive candidates for the synthesis of new redox active polymers. Monomer 5 depicted in Chart 2, involves the common β-linkage to thiophene to produce a tethered Ru5(bpy)2.21 Electrochemical deposition of this complex forms poly(Ru5(bpy)2) with saturated alkyl tethers that preclude the direct electronic communication between the metal complex and the conducting polymer backbone to give separate redox processes for the two electroactive components. A recent report also outlines the use of symmetrically disubstituted bipyridine ligand, 6, which possesses electropolymerizable groups tethered on both sides of the metal chelate to form metal complexes (iron and ruthenium) with multiple sites available for polymerization.22 In this case, the use of a terminal ethylene dioxythiophene, EDOT, group was employed to lower the anodic electropolymerization potential. These ligands and their metal complexes (Ru(6(X = S, n
= 4))(bpy)2, Ru(6(X = S, n
= 4))2Cl2, Fe(6(X = S, n
= 4))3) readily undergo electropolymerization to provide materials displaying electroactivity assigned to extended polythiophene (0.60 V) and immobilized metal complexes (0.35 V–Ru and 1.00 V–Fe)
Fig. 4.
 |
| | Fig. 4 Left: Potentiodynamic electropolymerization of Ru(6(X = S, n
= 4))2Cl2 from CH2Cl2 with 0.1 M Bu4NPF6 as supporting electrolyte by repeated scans at a rate of 100 mV s−1 (potential in V vs. Ag/AgCl). Growth of polymer film is indicated by sequential growth in current. Right: Cyclic voltammogram of the polymer film in a monomer-free electrolyte solution showing separate electrochemical responses for the two electroactive components (Adapted from ref. 22 by permission of The Royal Society of Chemistry). | |
 |
| | Chart 2 | |
Adjoined metal clusters and complexes
In addition to discreet metal complexes such as ferrocene or Ru(bpy)3, redox active organometallic metal clusters have been covalently attached to conducting polymer backbones. The work of Shin et al. has focused on exploring the electronic interactions between polythiophene backbones and various cobalt and molybdenum organometallic clusters attached through both conjugated and non-conjugated tethers.23–26 A selected example 7 is depicted in Chart 3 and the rich electrochemistry of the organometallic clusters provides potential applications including electrochromism. In a series of recent reports, Shin and coworkers outline the synthesis, characterization, and electrochemical properties of polythiophene/organometallic cluster hybrid materials including the electrodeposition of polymer films and their subsequent characterization by UV-Vis spectroelectrochemistry.23–26 The materials display defined and well-separated electrochemical responses, and the incorporation of the organometallic clusters results in significant perturbation of the polythiophene-based electrochromic response.
 |
| | Chart 3 | |
Mirkin and coworkers27,28 have reported the use of a terthiophene unit as a redox-switchable hemilabile ligand.29,30 In this system, a redox active ruthenium complex is covalently attached to the polymer backbone through an alkyldiphenylphosphine tether and a weaker Ru–S coordination interaction, 8, Chart 3. This unique architecture was designed to investigate if the electron donation to the metal center could be sequentially varied by adjusting the redox state of the polythiophene. These studies made use of FT-IR spectroelectrochemistry to probe electron density of the ruthenium metal center through changes in the characteristic carbonyl stretching frequency. Although, these studies demonstrate that charge localization on the metal centers prohibits the tuning of electronic properties over a continuous range, reversible small molecule uptake and release was achieved as a function of redox state, Chart 3.28
A novel class of phosphinoterthiophene ligands developed by Wolf and coworkers has been used to study the effects of incorporating a transition metal center directly into a conducting polymer framework.31–33 In this work, a series of terthiophene ligands were synthesized by appending a diphenylphosphine moiety to either the central or exterior ring of the terthiophene unit, a selected example is shown in Chart 3, monomer 9. The phosphinoterthiophene ligands direct palladium toward C-metallation and S-coordination to form a collection of metal complexes that differ in connectivity, morphology, and metal content. These complexes electropolymerize to form conductive electroactive thin films and EDX (energy-dispersive X-ray) analysis of the Pd/C, Pd/S, and Pd/Cl ratios of the polymer films confirm the retention of the monomer structure. The films display conductivities in the 10−4–10−3 S cm−1 range as measured by in situ conductivity with the use of interdigitated microelectrodes,34,35 and the authors conclude that the role of the metal is largely inductive. The ligands are hemilabile and UV-Vis and IR spectroscopy of these polymer films before and after treatment with isocyanide ligands that partially displace the palladium metal centers provide evidence as to the role of the transition metal centers.
Adjoined metal–salen complexes
Reynolds and coworkers have explored the synthesis, metal-binding, and electrochemical properties of a SALOTH (condensation product of salicylaldehyde and 3,4-diaminothiophene derivatives) ligand, 10, Chart 3.36–38 The Schiff-base ligands were chosen due to their ease of synthesis, high coordination affinity towards a variety of metal centers, and robust chemical nature. Studies of zinc, copper, nickel, and cobalt ions bound in the ligand and the effect on the ligand and polymer properties were investigated. This group's studies further reveal that the complexes can be polymerized between the para-positions of the SALOTH ligand to give a polyphenylene backbone or the α-positions of the thiophenes to produce a polythiophene backbone depending on the length of the thienyl portion and the substituent pattern on the SALOTH ligand. Specifically, if the thienyl portion of the ligand is extended to a terthiophene moiety and the para-positions of the SALOTH are capped with methyl groups a polythiophene backbone will be exclusively formed. Based on cyclic voltammetry and the electrochromic properties Reynolds and coworkers conclude that the metal centers inductively influence the electrochromic behavior of the polythiophene backbone.37,38
Inner sphere electronic coupling
In inner sphere conducting metallopolymers metal centers are part of the intrapolymer conductive pathway. This has been accomplished several different ways including: binding the metal centers to the backbone via metal–carbon or metal–heteroatom bonds, using the metal centers as functional linkers within the polymer backbone, using chelating ligands such as salen (N,N′-ethylenebis(salicylidenimine)) to introduce the metals, and more elaborate systems such as polymetallorotaxanes. After a discussion of the energetics, examples of each of these arrangements will be provided in the following sections.
Energy dispersion in metallopolymers
Although the phenomenon of redox matching in metal-containing conducting polymers has been increasingly discussed in the literature, we feel an important conceptual aspect of this electronic state has been overlooked to date. In order to take full advantage of the intimate communication between the transition metal centers and the conducting polymer backbone one must first consider the energetic effects of combining the two electro-active systems. In the case of molecular systems involving well-defined metal complexes such as ferrocene or Ru(bpy)3, there exists an isoenergic series of electronic states that can facilitate the charge migration through the system/solution. This is a direct result of the closed-shell, coordinatively saturated nature of the metal centers that limits the perturbations and leads to relatively sharp and cleanly reversible electrochemical transitions. This is graphically represented as seen in Fig. 5A. When these types of metal complexes are tethered to a polymer backbone which possesses its own band of electronic states, as in the outer sphere arrangement, the electronic states remain uniform, isoenergetic and distinct from the electronic states of the conducting polymer. One case of this environment is represented in Fig. 5B which would lead to reversible and well-separated electrochemistry for both the metal complexes and the conducting polymer backbone. Indeed, even a coincidental overlap of redox potentials still gives two redox systems that are independent of one another. The distinct disadvantage of such an arrangement lies in the coordinatively saturated nature of the metal complexes. The passive nature of the metal centers leads to well-behaved electrochemical responses, however this same trait prohibits utilizing direct interactions with the metal centers for sensing or catalytic applications.
 |
| | Fig. 5 Distribution of energetic states in molecular (top) and polymeric (bottom) systems. | |
In sharp contrast to this outer sphere electronic arrangement, the inner sphere arrangement enables the utilization of redox active metal centers that are coordinatively unsaturated as part of the conduction pathway. In a molecular system, the open coordination sphere of the metal centers leads to inherent sensitivity to the local environment due to ligand, solvent, and ionic interactions. The diverse and dynamic local environment is unique to each metal center and in turn creates an innate dispersion in the redox energies of the metal complexes as shown in Fig. 5C. This response is often averaged for molecular systems in solution but when confined to an electrode the disparity of local environments is evident. The result is a broad and varied metal centered electrochemical response. Although first consideration from an electrochemist's standpoint would deem this an inherent weakness, we maintain that, when carefully matched to a polymer system, the dispersion of states can be a powerful trait with many potential applications. More specifically, if a polymer system, and its broad distribution of electronic states, is overlaid on the dispersed metal-based electronic states in a judicious manner so as to match the disparity of the polymer system to the disparity in the metal system (Fig. 5D) one can obtain a redox-matched state that should be both highly conductive and sensitive to the local environments of the transition metal centers. Although there are relatively few examples of coordinatively unsaturated systems to date, the principles of energy dispersion are important to consider when designing metallopolymers for sensory and catalytic applications. However, the general principles of redox matching in metallopolymers outlined here are applicable to both coordinatively saturated and unsaturated systems to enhance electronic communication between the metal centers and organic segments of the metallopolymer.
As touched on in the introduction, a direct analogy between the polymeric systems discussed herein and molecular mixed valence systems can be drawn. The vast body of work on the molecular systems39–42 provides a valuable resource when considering the electronic interactions between metal centers within inner sphere metallopolymers.
Initial demonstration of redox matching
From a molecular design aspect, the salen ligand meets several important criteria, namely, it is: chemically robust, able to accommodate many different metals, easily synthesized, and capable of forming coordinatively unsaturated metal complexes. Our laboratory has made use of this versatile ligand to prepare a series of functional metallopolymers with high conductivities as summarized in Chart 4.43,44 Although the thiophene-free parent M(salen) system has been shown to oxidatively polymerize,45–52 the high voltages required to polymerize and dope the materials produces degraded metal complexes. Thiophene based electroactive groups have been incorporated para to the phenolic oxygens of the salen core in the monomers in order to lower the potential necessary to electropolymerize the salen–metal complexes and achieve the highest degree of communication between the polymer backbone and the metal centers. Initial cyclic voltammetry and in situ conductivity (max = 40 S cm−1) studies on 12 revealed that although the cobalt metal center was in direct communication with the polymer's electroactive π-system it was not involved in electron/hole transport, Fig. 6
(solid line).43 This conclusion was supported by the lack of overlap of the electrochemical responses for the cobalt and polymer backbone, and the loss of metal centered electroactivity in thick films due to a lack of facile Co+2/Co+3-based charge transport. Substitution of the thiophene moiety with an EDOT creates a more favorable redox match between the conducting polymer backbone and the transition metal center, Fig. 6
(dotted line), thus establishing a distribution of redox states similar to that shown graphically in Fig. 5D. This system has excellent organic–metal redox overlap that was commensurate with the onset of conductivity. The redox matching generated a large increase in the maximum conductivity to a value of 250 S cm−1, a value comparable to polyEDOT polymerized under the same conditions. It was concluded that the matching of the metal and polymer backbone centered redox potentials provides a conductivity enhancement beyond a simple additive combination. In order to clearly identify the role of the cobalt metal center in the conductivity, this system was reacted with a series of pyridine-based ligands that selectively perturbed the energy level of the metal and therefore affected the degree of redox coupling in the system. As shown in Fig. 7A, the redox couple of the cobalt centers shift to a lower (unmatched) potential while the polymer backbone was left unchanged. Additionally, Fig. 7B reveals that this led to a 66% decrease in the conductivity and an onset of conductivity that no longer involves the metal centers. The latter proves that the cobalt centers are electronically removed from the system while still being physically present. In this regard, pyridine also served as the initial demonstration of small molecule sensing via metallopolymer-modified electrodes in a redox-matched system.
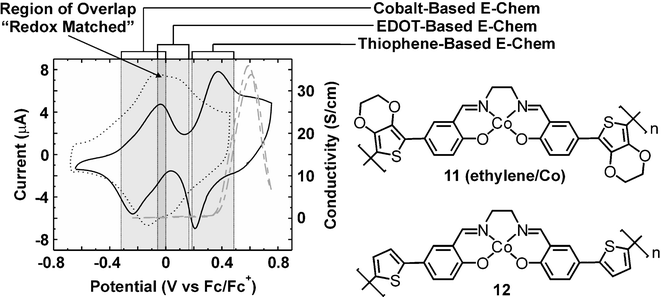 |
| | Fig. 6 Cyclic voltammogram of a thin film of polymer 12 showing well-separated redox potentials for the polymer backbone and the cobalt metal center (solid line). In situ conductivity versus electrochemical potential of polymer 12
(dashed gray line). The lack of redox matching in this system leads to relatively low conductivity. Cyclic voltammogram of a thin film of polymer 11
(ethylene bridge/Co)
(dotted line) showing overlapped or redox matched electrochemical processes. | |
 |
| | Fig. 7 A: Cyclic voltammogram of polymer 11(ethylene/Co) in 0.1 M Bu4NPF6/acetonitrile solution (solid line) and in the presence of pyridine (dotted line) demonstrating a shift of the metal-based peak as a result of metal–ligand interactions. B: In situ conductivity versus electrochemical potential of polymer 11(ethylene/Co) before (solid line) and after (dotted line) exposure to pyridine clearly showing a reduction of conductivity as a result of less favourable redox matching. (Adapted from ref. 43 by permission of Wiley-VCH publishing.) | |
 |
| | Chart 4 | |
In addition to the initial observation of conductivity enhancement by redox-matching in conducting metallopolymers, our group has reported a detailed study of the electronic properties of a series of salen-based metallopolymers, 11, where both the metal center and diamine backbone of the salen ligand were varied.44 This study utilized cyclic voltammetry, in situ conductivity, UV-Vis spectroelectrochemisty, and in situ EPR spectroscopy to elucidate the correlation between the interpolymer electronic communication, ligand structure and the transition metal center. Increased bulkiness of the diamine used to prepare the salen ligand gave decreased interchain communication and associated conductivity. This effect was also metal dependent with the redox inactive copper-based system exhibiting the largest conductivity decrease, and the redox-active nickel system was less sensitive. The variation between interchain and intrachain type mechanisms of electronic transport complicates conducting polymers relative to molecular systems.
Redox matched materials
Recent examples of redox matched conducting metallopolymers
Transition metal centers directly bound to the polymer backbone.
Organometallic heterocyclic polymers have been prepared by “metallacycling” polymerization reactions53,54 by Nishihara and coworkers, and both ruthenium and cobalt materials have been investigated.55,56 The soluble cobalt-containing polymer 13, synthesized by cyclization of the corresponding diacetylene and CpCo(PPh3), has the most favorable electrochemical properties and displays redox-conductivity of 10−4 S cm−1 when doped with I2.55 The authors postulated that the metal centers provide a conduit for charge transfer through the π-conjugated backbone and that a mixed valence state exists after chemical doping.
Pickup and coworkers57–61 have made extensive use of impedance spectroscopy, rotating disk voltammetry, and dual sandwich electrode voltammetry to elucidate charge transport mechanisms in several ruthenium- and osmium-containing conducting polymers. They outlined how the different conductivity mechanisms depend upon the nature of the interaction between the metal centers and the ligand backbone. The benzimidazole-based conducting polymer 14, Chart 5, has demonstrated strong polymer mediated communication between the metal centers.57–60 The pH dependent nature of the charge delocalization resulting from the protonation/deprotonation of the polymer backbone support its role in charge transport. These studies revealed the roles of redox matching in polymer-mediated charge transport and how superexchange type mechanisms depend on the degree of organic polymer–metal ion orbital overlap. In an effort to increase this orbital overlap Pickup and coworkers investigated polymer 15, Chart 5.61 In this system the electron rich poly(bithiophene-co-bithiazole) backbone was selected to lower the energy of the polymer HOMO to promote electronic communication with the metal center. Impedance spectroscopy confirmed the enhanced electron transfer rate due to increased organic polymer–metal ion electronic communication in polymer 15 relative to polymer 14. Moreover, impedance spectroscopy, see Fig. 8, further revealed that the closer redox matching in the ruthenium analog of 15 gave increased electron-transport over the osmium analog. Additional evidence for the involvement of the polymer backbone in the metal centered electronic conduction was obtained from a potentiometric charge transport analysis that showed a sharp drop in charge mobility as the polymer backbone was overoxidized to a form that does not efficiently mediate electron transport, Fig. 8.
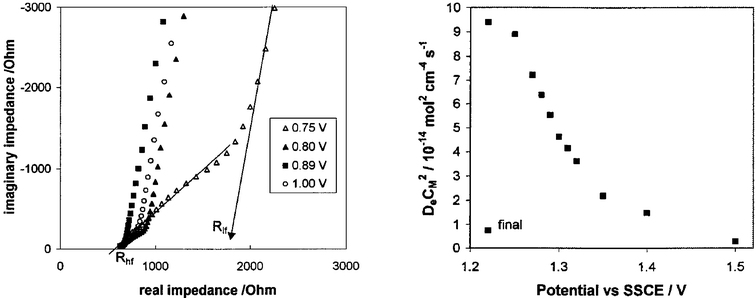 |
| | Fig. 8 Left: Complex plane impedance plots for a 15
(M = Os) coated Pt electrode at selected potentials in CH3CN containing Et4NClO4. Right: DeCM2versus potential from impedance data for a 15
(M = Ru) coated Pt electrode in CH3CN containing 0.1 M Et4NClO4. The order of experiments was from low (1.22 V) to high (1.5 V) potential, followed by a final experiment at 1.22 V. These experiments demonstrate the involvement of both the metal centers and the polymer backbone in charge transport through the polymeric material. (Adapted with permission from ref. 61. Copyright 2002 American Chemical Society.) | |
 |
| | Chart 5 | |
Our laboratory has also reported a series of polymeric systems based on the Ru(bpy)3 redox functionality.62,63 The most noteworthy material, in the context of this review, is the highly crosslinked poly16. Comparisons within a series of complexes demonstrated that the 4,4′-attachment of the bithiophene moieties to the bpy system provides the best inter-Ru(bpy)3 communication. This result is expected based upon the frontier orbitals of the bpy ligands and a high redox conductivity (3.3 × 10−3 S cm−1) was observed for poly16.63
Transition metal centers as part of the polymer backbone.
Recent studies by the research groups of Wolf and Higgins have focused on conducting polymers that incorporate ferrocene directly into the polymer backbone.64,65 Both groups have reported ferrocenes separated by oligothiophenes, 17 and 18, Chart 6, and although these studies do not report conductivity measurements, electrochemical and spectroelectrochemical data indicates that significant charge delocalization is present.66 Furthermore, studies in model systems show that a matching of the redox couples of the oligothiophene backbone to the ferrocene- greatly enhances the charge delocalization.66
 |
| | Chart 6 | |
Linear rod-like polymers based on oligothiophene functionalized terpyridine ligand systems have been reported.67–69 Charge mobility and polymer conductivity studies on polymer 19, Chart 668,69 determined that the combination of a bithiophene polymeric unit and the osmium–terpyridine redox-active complex gives ideal redox matching. The electrical transport properties were probed by a variety of electrochemical techniques including impedance spectroscopy and large amplitude potential step chronocoulometry. These techniques revealed a lower limit estimate of the conductivity of 0.5–1 × 10−4 S cm−1
(Fig. 9) that is enabled by the high degree of overlap between the quaterthiophene HOMOs and the orbitals of the osmium complex.68
 |
| | Fig. 9
In situ conductivity measurement using a 19-coated interdigiated platinum microelectrode array. (Adapted with permission from ref. 68. Copyright 2003 American Chemical Society.) | |
Polymetallorotaxanes and entwined metallopolymers.
Conducting polymers with metal-rotaxane and -entwined architectures provide access to interlocking network materials.70 The assembly of metallorotaxanes and entwined metallopolymers is templated by metal centers which serve to hold a circular molecular component in place about a linear molecule that pierces the macrocycle center and to hold two U-shaped molecules together (respectively). In the rotaxane variant the threading molecule is irreversibly captured by the macrocycle and in entwined metallopolymers the thread can be removed without breaking a covalent bond. Bidan, Divisia-Blohorn, Kern, Sauvage, and coworkers have published extensively on entwined conducting metallopolymers and polymetallorotaxane systems.71–78
In the case of the entwined metallopolymer systems,71 two ligands have been studied that incorporate thiophene-based electropolymerizable moieties in the 2,9-positions of the copper binding 1,10-phenanthroline ligands, 20 and 21, Chart 7. X-ray absorption spectroscopy of the monomer and polymer structures reveals that the copper coordination geometry remains unchanged in the polymer and depends on which ligand is used. Cyclic voltammetry of poly[Cu(20)2] reveals a poor overlap of the metal-centered and ligand-centered redox processes and an in situ conductivity of 1.1 × 10−4 S cm−1 which is similar to poly20
(1.2 × 10−4 S cm−1) suggesting no involvement of the copper ions in the conductivity. In contrast, polymers of alkylated terthiophene containing ligand, 21, provide better redox matching with the copper metal centers and an increase in the conductivity to 9 × 10−4 S cm−1. Again, the role of the metal can be inferred by comparing the metal-free polymer, poly21, which shows a decreased conductivity of 1 × 10−4 S cm−1 similar to the non-redox matched system.
 |
| | Chart 7 | |
Our laboratory reported the initial synthesis and characterization of conducting polymetallorotaxanes72,79 based on a 2,2′-bipyridyl threading element functionalized with polymerizable thienyl groups in the 5,5′-positions and Sauvage's 1,10-phenanthroline based macrocycle, structure 22, Chart 8. These architectures were designed to incorporate well-defined receptor sites into a conducting polymer framework that can reversibly bind transition metal ions. Studies on electropolymerized films showed that demetallation and remetallation processes are completely reversible leading to potential sensory applications and that the Lewis acidity and redox properties of different metal centers effected the polymer's electronic conductivity. Good redox matching was observed between the PEDOT based threading polymer and copper metal ions. In this system, a dramatic 106–107-fold increase in conductivity was observed upon treatment of the metal-free material with Cu+2 ions that effectively dope the polymer when they bind to the templated binding sites.72
 |
| | Chart 8 | |
Subsequent studies of polymetallorotaxane systems by Sauvage and coworkers73–77 focused on rotaxanes based upon the same macrocycle threaded with 2,9-functionalized 1,10-phenanthroline units (not shown).74,75,77 In contrast to 22, these materials can not be reversibly demetallated without the use of a lithium ion placeholder to preserve the rotaxane structure. A second-generation polymetallorotaxane structure, 23, Chart 8, repositions the electropolymerizable groups to the 3,8-positions of the phenanthroline resulting in a linear threading element and polymer backbone. Additional fine tuning of the properties was achieved by varying the substituents at the 2,9-positions.73,76 By incorporating more sterically demanding methyl substituents (as compared to hydrogens) the resulting polymer's electrochemical stability and metal binding reversibility are enhanced. Polymetallorotaxanes incorporating the 2,9-dimethyl phenanthroline 23 revealed that the charge-carrier mobilities were higher in the metal free forms than the lithium, cobalt, zinc, and copper metallorotaxanes. In polymer 23 redox matching is absent and the metal centers act as barriers to electronic conductivity due to charge localization mechanisms.
Through carefully designed synthesis and supramolecular assembly, our group has constructed a three-strand conducting metallorotaxane ladder polymer that incorporates dissimilar electroactive moieties into a superpolymer structure.78 As shown in Chart 9, polymer 24 has different electropolymerizable groups attached to both the macrocyclic and threading elements of the rotaxane structure. By introducing electroactive groups with distinctly different oxidation potentials these two groups can be polymerized in a step-wise fashion. Specifically, the EDOT-based groups covalently attached to the macrocycle can be first electropolymerized producing a metallorotaxane structure and the bithiophene groups polymerize at higher potentials. The size and relative orientation of the monomers in the second polymerization were chosen to produce a highly regular three-strand supramolecular ladder polymer. By including the two different types of conducting polymer backbones into one material, it was demonstrated that each can be electrochemically doped into its conductive state at a unique electrochemical potential creating a situation wherein the inner strand is conductive while the outer strands remain insulating; an insulated molecular wire. In this case, the copper ions that were redox matched to the insulated conducting polymer were key to interpolymer transport, see Fig. 10. Hence a conductivity of 38 S cm−1 was observed for the copper metallorotaxanes, whereas the analogous system incorporating electroinactive zinc ions exhibits a much lower conductivity of 2 S cm−1.
 |
| | Fig. 10 Conduction between molecular wires is mediated by metal-centered electroactivity. | |
 |
| | Chart 9 | |
Applications.
Several of the materials introduced in previous sections have been used in applications ranging from electroresponsive sensors to photoelectronic devices. In cases where the interaction of the transition metal centers with the conducting polymer backbones creates the relevant function; enhanced properties are obtained by redox matching. Selected examples of each application are briefly reviewed below.
Sensing.
We have illustrated how strong electronic coupling between the transition metal centers and the organic conducting polymer backbone facilitates electronic charge transport. The intimate involvement of the transition metal centers in the conductivity pathway provides an approach to chemoresistive chemical sensors. For example, the redox potential of a coordinatively unsaturated transition metal will be highly sensitive to small molecule or ion binding. Analyte induced changes in the metal's redox potential will affect the degree of overlap with the conducting polymer thereby creating a measurable response in conductivity. The matching of the metal center's and organic polymer's redox potentials can be either increased or decreased depending on whether the binding event shifts the redox potential of the metal center into a state of better overlap with the polymer backbone or worse overlap. These two scenarios lead to two distinct sensing strategies with characteristic levels of sensitivity that can be tuned and tailored depending on the desired application. As shown in Fig. 11, the most sensitive situation is achieved with a perfectly redox matched conducting metallopolymer. Here a single binding event can disrupt a critical transport pathway and lead to a drastic reduction in conductivity thereby providing an ultrasensitive system for detecting small molecules. Such a mechanism is best realized in systems that have a finite number of pathways to block. Alternatively, if the initial state of the conducting metallopolymer displays partial or no redox matching of the of the organic polymer and metal centers, a low conductivity charge localized material would be observed. In this situation, a single binding event can create favorable redox matching that will enhance local transport. However when the relative concentration of activated low resistivity regions exceeds a critical percolation level the bulk conductivity of the material will be enhanced leading to a measurable electrical output. The sensitivity of this motif is best suited to the detection of target analytes at intermediate concentrations. Initial, proof-of-concept, sensory systems based on conducting metallopolymers and the principles of variable redox matching within these systems are outlined in the following examples.
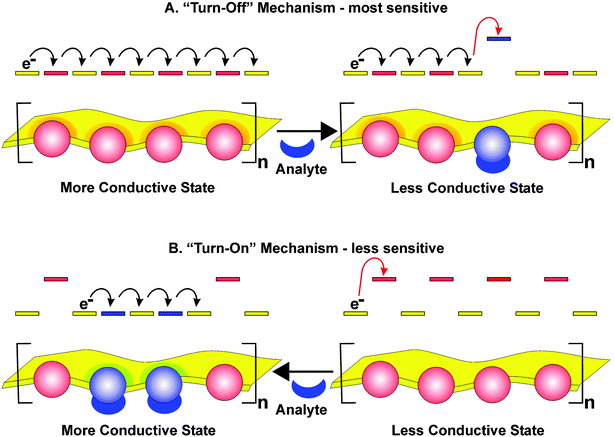 |
| | Fig. 11 Chemoresistive sensing schemes with conducting metallopolymers demonstrating how analyte induced energetic changes can produce a response. | |
Electropolymerization readily produces polymer-functionalized electrodes for the construction of resistivity/conductivity-based chemical sensors. One example, based on the polymetallorotaxane developed in our laboratory and discussed above, takes advantage of its reversible metal ion binding rotaxane structure, Chart 10
(25a and 25b).79 In particular these materials display changes in both their optical and electrical properties with metal coordination. Specifically, the addition of copper or zinc ions, gives a 34 nm red shift in the UV-Vis spectrum from the metal-free polymer, Fig. 12.
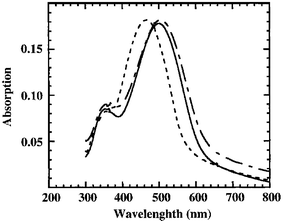 |
| | Fig. 12 Initial 25b
(M = Zn) film (solid line), after treatment with H2O/ethylenediamine (3 ∶ 1)
(dashed line) and after dipping in 0.05 M Zn(ClO4)2/CH3CN (dot-dashed line). Reversible metal binding causes 34 nm shift in UV-Vis spectrum of polymer film. (Adapted with permission from ref. 79. Copyright 1996 American Chemical Society.) | |
 |
| | Chart 10 | |
Conducting metallopolymers based upon the salen ligand system are particularly well-suited for sensory applications as a result of the nominally vacant axial sites that enable further coordination to specific analytes. Our group recently reported the use of a cobalt-based salen polymer, 26, as a resistivity-based detector for nitric oxide.80 In this system it had been previously observed that the cobalt-based redox processes provided good, but not optimal, redox matching with the organic polymer backbone, thereby establishing a turn-on sensor system (Fig. 11). When exposed to a 7 mM solution of NO, the electrode confined polymer exhibits a specific 30% increase in its in situ conductivity. This increase is accompanied by an anodic shift of the cobalt-centered redox activity, which enhances the redox matching with the organic polymer. The sensor demonstrated a highly reversible response by returning to the initial state within three potential sweeps in an NO-free solution. Additionally, this sensor system has shown good stability in the aerobic aqueous conditions opening potential for use in biological media.
Another example utilizing a metal-containing conducting polymer in a sensory role was demonstrated by Reynolds and coworkers with the crown-ether containing polymer, 27, Chart 10.81 Once electrodeposited on an electrode surface this polymer has two “hot-spots” that can be utilized for the detection of different classes of analytes. First, the chelated metalloSALOTH unit, which is in close electronic communication with the intramolecular conduction pathway, can detect neutral donor molecules (pyridine, DMF, DMAc, DMSO) down to nM concentrations by virtue of semi-reversible (50% recovery) disappearance of the electroactivity of the polymer film. Second, the tethered crown ether moiety shows cathodic electrochemical shifts in electroactivity in response to Li, Na, Mg, and Ba ions. The monocations of Li and Na show fully reversible binding behavior whereas dicationic Mg and Ba bind irreversibly in the crown ether.
Electrocatalysis.
Transition metal centers in a conducting polymer framework have many potential uses in catalytic processes including: tuning the reactivity of catalysts based on applied voltage, driving reactions based on electrical switching of the transition metal centers redox state, and delivering multiple electrons to a reaction site for a single transformation. An example of the latter is the four-electron electrocatalytic reduction of oxygen to water. The same polymeric material utilized for the nitric oxide detection has been shown to efficiently reduce oxygen to water, 26, Chart 10.82 The high conductivity due to the redox matching allows the rapid delivery of electrons to the cobalt and the complete conversion of the oxygen to water with almost no trace of hydrogen peroxide formation being detected by rotating disk voltammetry. In contrast the parent poly(Co/salen) system with no redox matching and lower conductivity gives only a 39% conversion.
Photoelectronic devices.
The strong charge-transfer UV-Visible absorptions of many transition metal complexes are attractive for incorporation into photonic devices. When this feature is combined with a conducting polymer backbone that can efficiently shuttle charge between the metal centers, photoelectronic applications can be easily imagined. One such device makes use of alternating layer by layer deposition of conducting metallopolymer 28 and SPAN, Chart 11,83 to create a photodetector based upon charge injection into a conducting polymer from excited state of the ruthenium metal center. Under simulated solar illumination, functioning devices have exhibited short circuit currents and open circuit voltages in the 8.9–15.0 µA cm−2 and 0.76–0.84 V ranges (respectively). These promising results and the ability to tune the absorption of the device by introducing different transition metal complexes suggests a promising future for conducting metallopolymers in photoelectronics.
 |
| | Chart 11 | |
Concluding remarks
In this review we have provided a general introduction to the principles governing redox active transition metal centers in organic conducting polymer systems. The resulting perturbations can profoundly effect the conductivity and several examples were provided from the recent literature. Additionally we have outlined how the redox matching of the metal centered electroactivity with that of the electroactive organic polymer backbone can be used to enhance conductivity, sensory responses, and electrocatalytic activity. Due to the relatively young nature of the field of highly conducting metallopolymers the potential of such systems remains largely unexploited. Future work in this area will undoubtedly provide novel materials with major impact on the fields of sensory devices, electrocatalysis, optoelectronics, energy conversion and storage, and molecular electronics.
Acknowledgements
We would like to acknowledge the U.S. Army through the Institute for Soldier Nanotechnologies, under Contract DAAD-19-02-D-002 with the U.S. Army Research Office. The content does not necessarily reflect the position of the Government, and no official endorsement should be inferred.
References
- H. Shirakawa, Angew. Chem. Int. Ed., 2001, 40, 2574–2580 CrossRef.
-
Handbook of Conducting Polymers, Second Edition, eds. T. A. Skotheim, R. L. Elsenbaumer and J. R. Reynolds, 1997 Search PubMed.
-
J. D. Atwood, Inorganic and Organometallic Reaction Mechanisms, Wiley, New York, 1997 Search PubMed.
-
G. E. Rodgers, Introduction to Coordination, Solid State, and Descriptive Inorganic Chemistry, McGraw-Hill, New York, 1994 Search PubMed.
-
I. Manners, Synthetic Metal-Containing Polymers, Wiley-VCH, Weinheim, Germany, 2004 Search PubMed.
- P. Nguyen, P. Gómez-Elipe and I. Manners, Chem. Rev., 1999, 99, 1515–1548 CrossRef CAS.
- I. Manners, Science, 2001, 294, 1664–1666 CrossRef CAS.
- U. S. Schubert and C. Eschbaumer, Angew. Chem. Int. Ed., 2002, 41, 2892–2926 CrossRef CAS.
- M. O. Wolf, Adv. Mater., 2001, 13, 545–553 CrossRef CAS.
- J. Roncali, J. Mater. Chem., 1999, 9, 1875–1893 RSC.
- M. O. Wolf and Y. Zhu, Adv. Mater., 2000, 12, 599–601 CrossRef CAS.
- R. P. Kingsborough and T. M. Swager, Transition Metals in Polymeric π-Conjugated Organic Frameworks, Prog. Inorg. Chem., 1999, 48, 123–231 CAS.
- C. G. Cameron, B. J. MacLean and P. G. Pickup, Macromol. Symp., 2003, 196, 165–171 CrossRef CAS.
- P. G. Pickup, J. Mater. Chem., 1999, 9, 1641–1653 RSC.
- T. L. Stott and M. O. Wolf, Coord. Chem. Rev., 2003, 246, 89–101 CrossRef CAS.
- R. Back and R. B. Lennox, Langmuir, 1992, 8, 959–964 CrossRef CAS.
- G. Zotti, S. Zecchin, G. Schiavon, A. Berlin, G. Pagani and A. Canavesi, Chem. Mater., 1995, 7, 2309–2315 CrossRef CAS.
- G. Zotti, G. Schiavon, S. Zecchin, A. Berlin, G. Pagani and A. Canavesi, Synth. Met., 1996, 76, 255–258 CrossRef CAS.
- G. Zotti, G. Schiavon, S. Zecchin, A. Berlin, A. Canavesi and G. Pagani, Synth. Met., 1997, 84, 239–240 CrossRef CAS.
- H. Brisset, A.-E. Navarro, C. Moustrou, I. F. Perepichka and J. Roncali, Electrochem. Commun., 2004, 6, 249–253 CrossRef CAS.
- J. A. Crayston, A. Iraqi, J. J. Morrison and J. C. Walton, Synth. Met., 1997, 84, 441–442 CrossRef CAS.
- B. Jousselme, P. Blanchard, M. Oçafrain, M. Allain, E. Levillain and J. Roncali, J. Mater. Chem., 2004, 14, 421–427 RSC.
- D. H. Kim, B. S. Kang, S. M. Lim, K.-M. Bark, B. G. Kim, M. Shiro, Y.-B. Shim and S. C. Shin, J. Chem. Soc., Dalton Trans., 1998, 1893–1898 RSC.
- B. S. Kang, D. H. Kim, T. S. Jung, E. K. Jang, Y. Pak, S. C. Shin, D.-S. Park and Y.-B. Shim, Synth. Met., 1999, 105, 9–12 CrossRef CAS.
- D. H. Kim, D.-S. Park, Y.-B. Shim and S. C. Shin, J. Organomet. Chem., 2000, 608, 133–138 CrossRef CAS.
- D. H. Kim, J.-H. Kim, T. H. Kim, D. M. Kang, Y. H. Kim, Y.-B. Shim and S. C. Shin, Chem. Mater., 2003, 15, 825–827 CrossRef CAS.
- D. A. Weinberger, T. B. Higgins, C. A. Mirkin, L. M. Liable-Sands and A. L. Rheingold, Angew. Chem. Int. Ed., 1999, 38, 2565–2568 CrossRef CAS.
- D. A. Weinberger, T. B. Higgins, C. A. Mirkin, C. L. Stern, L. M. Liable-Sands and A. L. Rheingold, J. Am. Chem. Soc., 2001, 123, 2503–2516 CrossRef CAS.
- C. S. Slone, D. A. Weinberger and C. A. Mirkin, Prog. Inorg. Chem., 1999, 48, 233–350 CAS.
- A. Bader and E. Lindner, Coord. Chem. Rev., 1991, 108, 27–110 CrossRef CAS.
- O. Clot, M. O. Wolf and B. O. Patrick, J. Am. Chem. Soc., 2000, 122, 10456–10457 CrossRef CAS.
- O. Clot, M. O. Wolf and B. O. Patrick, J. Am. Chem. Soc., 2001, 123, 9963–9973 CrossRef CAS.
- O. Clot, Y. Akahori, C. Moorlag, D. B. Leznoff, M. O. Wolf, R. J. Batchelor, B. O. Patrick and M. Ishii, Inorg. Chem., 2003, 42, 2704–2713 CrossRef CAS.
- G. P. Kittlesen, H. S. White and M. S. Wrighton, J. Am. Chem. Soc., 1984, 106, 7389–7396 CrossRef CAS.
- It is important to note that comparisons of absolute conductivity measurements made by this or other techniques between materials are difficult to standardize due to variations in sample handling and conditions. However, when the experiments are self-consistent and properly calibrated the general trends observed are meaningful.
- J. L. Reddinger and J. R. Reynolds, Synth. Met., 1997, 84, 225–226 CrossRef CAS.
- J. L. Reddinger and J. R. Reynolds, Macromolecules, 1997, 30, 673–675 CrossRef CAS.
- J. L. Reddinger and J. R. Reynolds, Chem. Mater., 1998, 10, 1236–1243 CrossRef CAS.
- M. B. Robin and P. Day, Adv. Inorg. Chem. Radiochem., 1967, 10, 247–422 CAS.
- D. E. Richardson and H. Taube, Coord. Chem. Rev., 1984, 60, 107–129 CrossRef CAS.
- D. E. Richardson and H. Taube, J. Am. Chem. Soc., 1983, 105, 40–51 CrossRef CAS.
- B. S. Brunschwig, C. Creutz and N. Sutin, Chem. Soc. Rev., 2002, 31, 168–184 RSC.
- R. P. Kingsborough and T. M. Swager, Adv. Mater., 1998, 10, 1100–1104 CrossRef CAS.
- R. P. Kingsborough and T. M. Swager, J. Am. Chem. Soc., 1999, 121, 8825–8834 CrossRef CAS.
- L. A. Hoferkamp and K. A. Goldsby, Chem. Mater., 1989, 1, 348–352 CrossRef.
- K. A. Goldsby, J. K. Blaho and L. A. Hoferkamp, Polyhedron, 1989, 8, 113–115 CrossRef CAS.
- K. A. Goldsby, J. Coord. Chem., 1988, 19, 83–90 CAS.
- P. Audebert, P. Capdevielle and M. Maumy, New J. Chem., 1992, 16, 697–703 Search PubMed.
- P. Audebert, P. Capdevielle and M. Maumy, Synth. Met., 1991, 43, 3049–3052 CrossRef CAS.
- P. Audebert, P. Capdevielle and M. Maumy, New J. Chem., 1991, 15, 235–237 Search PubMed.
- C. E. Dahm and D. G. Peters, Anal. Chem., 1994, 66, 3117–3123 CrossRef CAS.
- F. Bedioui, E. Labbe, S. Gutierrez-Granados and J. Devynck, J. Electroanal. Chem., 1991, 301, 267–274 CrossRef CAS.
- I. Tomita, A. Nishio, T. Igarashi and T. Endo, Polym. Bull., 1993, 30, 179–186 CrossRef CAS.
- J. R. Nitschke, S. Zürcher and T. D. Tilley, J. Am. Chem. Soc., 2000, 122, 10345–10352 CrossRef CAS.
- H. Nishihara, M. Kurashina and M. Murata, Macromol. Symp., 2003, 196, 27–38 CrossRef CAS.
- M. Kurashina, M. Murata, T. Watanabe and H. Nishihara, J. Am. Chem. Soc., 2003, 125, 12420–12421 CrossRef CAS.
- C. G. Cameron and P. G. Pickup, Chem. Commun., 1997, 303–304 RSC.
- C. G. Cameron and P. G. Pickup, J. Am. Chem. Soc., 1999, 121, 7710–7711 CrossRef CAS.
- C. G. Cameron, T. J. Pittman and P. G. Pickup, J. Phys. Chem. B, 2001, 105, 8838–8844 CrossRef CAS.
- C. G. Cameron and P. G. Pickup, J. Am. Chem. Soc., 1999, 121, 11773–11779 CrossRef.
- B. J. MacLean and P. G. Pickup, J. Phys. Chem. B, 2002, 106, 4658–4662 CrossRef CAS.
- S. S. Zhu and T. M. Swager, Adv. Mater., 1996, 8, 497–500 CAS.
- S. S. Zhu, R. P. Kingsborough and T. M. Swager, J. Mater. Chem., 1999, 9, 2123–2131 RSC.
- Y. Zhu and M. O. Wolf, Chem. Mater., 1999, 11, 2995–3001 CrossRef CAS.
- S. J. Higgins, C. L. Jones and S. M. Francis, Synth. Met., 1999, 98, 211–214 CrossRef.
- Y. Zhu and M. O. Wolf, J. Am. Chem. Soc., 2000, 122, 10121–10125 CrossRef CAS.
- J. Hjelm, E. C. Constable, E. Figgemeier, A. Hagfeldt, R. Handel, C. E. Housecroft, E. Mukhtar and E. Schofield, Chem. Commun., 2002, 284–285 RSC.
- J. Hjelm, R. W. Handel, A. Hagfeldt, E. C. Constable, C. E. Housecroft and R. J. Forster, J. Phys. Chem. B, 2003, 107, 10431–10439 CrossRef CAS.
- J. Hjelm, R. W. Handel, A. Hagfeldt, E. C. Constable, C. E. Housecroft and R. J. Forster, Electrochem. Commun., 2004, 6, 193–200 CrossRef CAS.
- J.-M. Kern, J.-P. Sauvage, G. Bidan and B. Divisia-Blohorn, J. Polym. Sci. Polym. Chem., 2003, 41, 3470–3477 Search PubMed.
- P.-L. Vidal, B. Divisia-Blohorn, G. Bidan, J.-L. Hazemann, J.-M. Kern and J.-P. Sauvage, Chem. Eur. J., 2000, 6, 1663–1673 CrossRef CAS.
- S. S. Zhu and T. M. Swager, J. Am. Chem. Soc., 1997, 119, 12568–12577 CrossRef CAS.
- B. Divisia-Blohorn, F. Genoud, C. Borel, G. Bidan, J.-M. Kern and J.-P. Sauvage, J. Phys. Chem. B, 2003, 107, 5126–5132 CrossRef CAS.
- P. L. Vidal, M. Billon, B. Divisia-Blohorn, G. Bidan, J. M. Kern and J. P. Sauvage, Chem. Commun., 1998, 629–630 RSC.
- P. L. Vidal, B. Divisia-Blohorn, M. Billon, G. Bidan, J. M. Kern and J. P. Sauvage, Synth. Met., 1999, 102, 1478–1479 CrossRef CAS.
- J.-P. Sauvage, J.-M. Kern, G. Bidan, B. Divisia-Blohorn and P.-L. Vidal, New J. Chem., 2002, 26, 1287–1290 RSC.
- P.-L. Vidal, B. Divisia-Blohorn, G. Bidan, J.-M. Kern, J.-P. Sauvage and J.-L. Hazemann, Inorg. Chem., 1999, 38, 4203–4210 CrossRef CAS.
- J. Buey and T. M. Swager, Angew. Chem. Int. Ed., 2000, 39, 608–612 CrossRef CAS.
- S. S. Zhu, P. J. Carroll and T. M. Swager, J. Am. Chem. Soc., 1996, 118, 8713–8714 CrossRef CAS.
- T. Shioya and T. M. Swager, Chem. Commun., 2002, 1364–1365 RSC.
- J. L. Reddinger and J. R. Reynolds, Chem. Mater., 1998, 10, 3–5 CrossRef CAS.
- R. P. Kingsborough and T. M. Swager, Chem. Mater., 2000, 12, 872–874 CrossRef CAS.
- K. Y. K. Man, H. L. Wong, W. K. Chan, C. Y. Kwong and A. B. Djurišić, Chem. Mat., 2004, 16, 365–367 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2005 |
Click here to see how this site uses Cookies. View our privacy policy here.