Robotic sequential analysis of a library of metalloporphyrins as electrocatalysts for voltammetric nitric oxide sensors
Victoria
Ryabova
,
Albert
Schulte
,
Thomas
Erichsen
and
Wolfgang
Schuhmann
*
Anal. Chem. – Elektroanalytik & Sensorik, Ruhr-Universität Bochum, Universitätsstraße 150, D-44780 Bochum, Germany. E-mail: wolfgang.schuhmann@rub.de; Fax: +49-234-3214683; Tel: +49-234-3226200
First published on 13th July 2005
Abstract
A library of 83 metalloporphyrins with varying substitution pattern at the meso-position of the porphyrins and different central metal ions in the core region has been synthesized in small quantities using a parallel synthesis strategy. By means of a specially designed electrochemical robotic device integrating a 96-well microtitre plate and an easily movable assembly of working, counter and reference electrodes, the different porphyrins were automatically applied in sequence to an in-well electrochemical preparation and testing of NO sensors. Screening the entire compound collection suggested initial considerations concerning the influence of varied functionalities of the metalloporphyrins on their electrocatalytic properties for the oxidation of NO and helped to identify the quality of the investigated catalyst candidates. As compared to manually performed quality tests, the proposed strategy of automation has advantages in convenience, rapidity and especially reproducibility avoiding any inaccuracies introduced by manually performing all steps of the complex sensor formation and testing sequence.
Introduction
Nitric oxide (NO), a small and structurally rather simple inorganic molecule, is synthesized by specialized mammalian cells and is known to play a double-faced regulatory role in the living organism, being a chemical messenger controlling important physiological functions as well as a cytotoxic agent involved in a number of pathological processes.1 The free radical NO gas has been identified to be the endothelial-derived relaxing factor (EDRF), which is released from endothelial cells in the blood vessels and governs vasodilatation. Additionally, NO functions as a fast-diffusing neurotransmitter in the central nervous system, where it participates in interneuronal communication, the control of cerebral blood flow, the modulation of synaptic plasticity and memory formation. Last but not least, NO is closely associated with prevention of platelet aggregation and is employed by the immune system either directly or through intracellular signalling as an effective initiator of host defence against tumour cells and infectious pathogens. As expected from the numerous roles that NO has, abnormalities in concentrations of endogenous NO due to inappropriate production and/or dysfunctions in metabolism in relevant active zones are highly critical for the state of health and have been implicated in common ailments such as stroke, ischemia, cardiac failure, hypertension, diabetes mellitus, or CNS disorders such as Huntington's, Parkinson's and Alzheimer's diseases.Because of the diversity of its functional repertoire, elucidating the mechanisms of NO action in the body has become a vigorous field of research in medicine and biology. However, the reactive and thus short-lived nature of the analyte, low physiologically relevant concentrations in the nanomolar range and the complexity of biological matrices make exceptionally sensitive and selective micro analytical methods an unequivocal prerequisite for monitoring dynamic changes in local concentrations of NO in vivo or in vitro. Colorimetric, fluorometric, spectroscopic and electrochemical methods were established for NO detection in biological samples,2 but of these only amperometry and voltammetry proved to be practicable for directly quantifying natural trace levels of NO in situ under physiological conditions. Chemically modifying the active tips of voltammetric noble metal or carbon microelectrodes with appropriate catalysts for the anodic oxidation of NO is the key to obtaining sensors with a sufficiently sensitive current response for the small NO concentration transiently appearing in tissue and close to cultured cells.3–7 Based on the pioneering work of Malinski and Taha,8 metalloporphyrins9–14 have been proposed as effective electrocatalysts in NO microsensors. Subsequently, metallated salens,12,15 and metallophthalocyanines16,17 have been demonstrated to be suitable electrocatalysts for NO oxidation. Despite the fact that several of these metallo-organic complexes with open coordination sites for the intermediate binding of NO have already been identified as suitable electrode modifiers, no clear trend is yet available linking the molecular chemical structure of a substance with its electrocatalytic activity and suggesting a more rational approach towards advanced NO-oxidizing catalysts. Quite the opposite, the development of NO electrocatalysts currently seems to depend on an Edisonian strategy of trial and error, with no robust basis and/or bottom-up theoretical guidance for predicting their value a priori.
A methodical study of the roles played by different structural components in the effectiveness of metallo-organic catalysts for the oxidation of NO would require screening of many compounds with a wide range of peripheral substituents and central metal atoms. Performing deterministic quality checks manually on a considerable number of candidates would be tedious and particularly time consuming. Under these circumstances, a combinatorial approach coupled with computerized electrochemical sensor tests should be more suitable for systematically assessing the properties of a complex set of e.g. metalloporphyrin-type NO catalysts with an enhanced throughput. Combinatorial chemistry is addressing parallel and automated synthesis and/or analysis and is nowadays widely used in modern research and development as a high-throughput technique for rapid drug discovery18–24 and optimization of heterogeneous catalysts.25–28 In electrochemistry, however, combinatorial schemes only recently became valuable tools for fabricating and screening libraries of functionalised conducting polymers,29–32 engineered metalloproteins,33,34 redox storage35 or ion sensing36 materials, and electrocatalysts for batteries,37–39 fuel cells40–44 or enzyme-based amperometric biosensors.45–47
Here, we present a study on the adaptation of an electrochemical robotic screening device46,48,49 for automatically assessing the electrocatalytic properties of metalloporphyrin derivatives with respect to the anodic oxidation of NO. A collection of more than 80 metalloporphyrins was parallely synthesized under variation of the substitution pattern at the meso-position of the porphyrins and of the central metal ion. The organization of the small compound library in the wells of a microtitre plate together with successful operation of a movable assembly of the working, counter and reference electrodes in the small volumes of the individual wells allowed the robotic system to apply the different metalloporphyrins one after another to an in-well electrochemical preparation and testing of NO sensors. The results of the robotic survey showed a pronounced influence of the varied functionalities of the metalloporphyrins on their electrocatalytic properties with respect to NO oxidation. As will be demonstrated, the proposed strategy of automatic sequential analysis has a high potential to decrease the time and labour associated with sensor screening, to facilitate the identification of good and elimination of poor electrocatalyst candidates, and to improve the repeatability and reproducibility of the test procedure avoiding any errors imposed by manually performing the complex sensor fabrication and evaluation sequence.
Experimental
Reagents
Nickel(II)tetrakis(3-methoxy-4-hydroxyphenyl)porphyrin was purchased from Porphyrin Systems (Lübeck, Germany). All benzaldehydes, trifluoroboron etherate (BF3·OEt2), tetraphenylchlorophosphate (Ph4PCl), sodium nitrite, sodium hydroxide, potassium ferrocyanide, and sulfuric acid were obtained from Merck (Darmstadt, Germany) and used as received. 2,3-Dichloro-5,6-dicyano-p-benzoquinone, N,N-dimethylformamide, pyrrole, and dichloromethane were purchased from Sigma-Aldrich (Steinheim, Germany). The latter three compounds were distilled for purification prior to use. Phosphate buffer solutions (PBS) were prepared with tri-distilled water using K2HPO4/KH2PO4 from Sigma-Aldrich Chemie (Steinheim, Germany). Metalloporphyrins were synthesized with reagent grade iron(II) chloride tetrahydrate, copper(II) chloride dihydrate, cobalt(II) acetate tetrahydrate, nickel(II) chloride tetrahydrate and zinc(II) chloride, all from Sigma-Aldrich Chemie (Steinheim, Germany).Instrumentation of the electrochemical robotic device
The main components of the electrochemical robotic device were three computer-controlled positioning devices (Linear Measuring Stages Limes 90, Owis, Staufen, Germany). One of them was equipped with an electrode holder and used for vertical (z) movements of a three-electrode assembly. The other two were combined and allowed horizontal (x, y) movement of 96-well glass or Teflon microtitre plates (wells in rows 1–12 and columns A–H; Zinsser Analytic, Frankfurt, Germany) that were placed on an attached platform.Platinum (Pt) disk working electrodes (r = 125 µm) were fabricated by heat-sealing 250 µm diameter Pt wires (Goodfellow, Bad Nauheim, Germany) into slightly tapered endings of pulled borosilicate glass capillaries and subsequently exposing a smooth Pt disk through careful polishing with emery paper and alumina paste. The Pt working electrode was assembled with a 250 µm diameter Pt wire serving as counter electrode and a 500 µm diameter Ag wire used as pseudo reference electrode in such a way that the working electrode was about 5 mm longer than the other two electrodes. This helped to avoid any contamination of the reference and counter electrodes with metalloporphyrin deposits while dip-coating the working electrode in the porphyrin solutions for sensor fabrication.
Cyclic voltammetry (CV) and differential pulse voltammetry (DPV) were carried out with the voltammetric analysis system E 611 from Metrohm (Herisau, Switzerland) in solutions that contained potassium ferrocyanide as an internal standard. A PC in combination with Windows software programmed in Microsoft Visual Basic 3.0 (Microsoft, Unterschleißheim, Germany) was in command of the positioning devices, controlled the automatically performed sequences of electrochemical measurements, and was used for data acquisition.
Synthesis of metalloporphyrins and preparation of NO solutions
A one-flask synthesis of porphyrins with different substitution patterns on their skeleton followed a standard literature protocol50 and was performed separately, in parallel, in multiple reaction vessels. In brief, 20 screw-capped V-vials for small-scale organic reactions (Sigma-Aldrich Chemie, Steinheim, Germany) were charged with tiny magnetic stirrers and 100 µmol of one of the 20 different para-substituted benzaldehydes, 1 mL of a solution containing 31.68 µL of BF3·OEt2 (250 µmol) and 2.8 mg of Ph4PCl (7.5 µmol) in 25 mL dichloromethane and finally 7.0 µL (100 µmol) of pyrrole. Subsequent to stirring for 30 min at room temperature, 19.3 mg 2,3-dichloro-5,6-dicyano-p-benzoquinone (85 µmol) were added as oxidizing agent and the mixture was stirred for another two hours. After completion of the reactions, the crude products were loaded on a neutral aluminium oxide column (3 cm, activity grade I) with 20 mL of dichloromethane as an eluent to extract the porphyrins. The solvent was evaporated, the porphyrins dissolved in 5 mL of N,N-dimethylformamide and the obtained solutions divided into portions of 1 mL. Metallation was then achieved simply by mixing the porphyrin solutions with stoichiometric quantities of the divalent metal salts of choice and keeping the resulting reaction mixtures (20 porphyrins × 5 metals) for 2 hours at 100 °C. Successful formation of the porphyrins and their metallated counterparts was confirmed spectroscopically by recording UV-vis absorption spectra and inspecting the typical Soret and Q-bands that are characteristic of free-base porphyrins and shift upon metallation.Nitric oxide was generated under rigorous exclusion of molecular oxygen using a closed argon/vacuum line by dropping 1 M sulfuric acid into 2 M sodium nitrite. It was allowed to pass through a saturated NaOH solution for purification and then bubbled into 5 mL of PBS at +4 °C and pH 7 for a minimum of 30 minutes to ensure saturation. Until use, the NO stock solution was stored at +4 °C in a tight-capped vial with a septum inlet. The final concentration of NO was determined using the Griess test to be about 3.5 mM at +4 °C.
Automated in-well electrochemical preparation and testing of NO sensors
To permit sequential measurements in an oxygen-free environment, the platform holding the 96-well microtitre plate was placed inside a rectangular Teflon container, which had an in- and outlet for argon at the side and was softly covered by a glass plate, forming an airtight seal. The lid had a hole just large enough to easily insert the three-electrode assembly and a metal needle, which was coupled via a PFA (perfluoroalkoxy) tubing, which is not permeable to molecular oxygen, to a stepmotor-actuated syringe pump (Hamilton, Martinsried, Germany) and used to deliver defined volumes of the NO stock solution into chosen wells. Moving the electrode assembly up and down and displacing the microtitre plate in the x, y direction were automated steps for sequentially approaching individual wells of the microtitre plate with the electrodes. For the in-well preparation of the metalloporphyrin-based NO sensors and the automatic screening of their electrocatalytic performance, 96-well microtitre plates were filled with deoxygenated solutions of: DMF (200 µL, A1–A12 and E1–E12), 10 of the metalloporphyrins dissolved in DMF (50 µL, row B2–B11), nickel(II)-tetrakis(3-methoxy-4-hydroxyphenyl)porphyrin dissolved in DMF (50 µL, row B12, used as reference), PBS, pH 7 (200 µL, row C1–C12), 10 mM ferrocyanide in PBS, pH 7 (100 µL, row D1–D12), water (200 µL, F1–F12 and H1–H12), 1 M sulfuric acid (200 µL, G1–G12).Results and discussion
The electrochemical detection of NO involves the oxidation of the molecule at the surface of an electrode polarized at a sufficiently high potential and the measurement of the related oxidation current. One metalloporphyrin successfully employed previously to promote the catalytically enhanced oxidation of NO at chemically modified electrodes was Ni(II)tetrakis(3-methoxy-4-hydroxyphenyl)porphyrin (the so-called “Malinski–Taha”, MT-type, porphyrin).8 Later, other porphyrins complexed with metals such as Mn(II), Co(II) and Pd(II) were also proven to be suitable electrode modifiers for the construction of sensitive NO sensors.9–14 However, although a large number of different metalloporphyrins have already been used for the construction of NO sensors, to the best of our knowledge no attempts have been made to elucidate the influence of the peripheral substituents and the central metal atoms on the electrocatalytic activity of different metalloporphyrin systems. Obviously, attempts in this direction need a large variety of similar metalloporphyrins and a reliable analytical procedure to generate sufficient data for elucidating trends in the complex parameter space. This in mind, a library of metalloporphyrins was synthesized with various central metal ions and substitution patterns at the meso-position of the porphyrin ring. By means of a parallel synthetic approach, pyrrole was allowed to react with equimolar quantities of 20 different substituted benzaldehydes to form a set of porphyrins each carrying four identically substituted phenyl groups in the meso-positions of the macrocyclic tetrapyrrole ring system. As depicted in Scheme 1, substituents with either electron-donating or -withdrawing character were chosen for the meso-phenyl groups thus influencing the electron density of the macrocyclic aromatic ring system, the redox potential of the metalloporphyrin, and concomitantly modulating the binding of the NO radical to the coordination site at the centre of the metallated macrocyle. These variations should have a pronounced impact on the electrocatalytic properties of the metalloporphyrin concerning NO oxidation.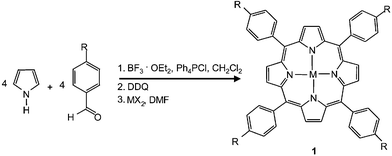 | ||
| Scheme 1 Reaction scheme as used for the synthesis of a library of metalloporphyrins with varying substituents (R) at the phenyl groups in the meso-positions of the porphyrin ring system and with 5 different central metal ions inserted into the core region. (R = H, Me, 2,4,6-Me, 2,3,4,5,6-Me, Et, Pr, OMe, OEt, OBu, NMe2, NEt2, OH, SMe, F, Cl, Br, CN, NO2, CF3, OCF3; MX2 = FeCl2, Co(OOCCH3)2, NiCl2, CuCl2, ZnCl2. | ||
Complexation of the obtained porphyrins with five transition metals from the fourth period of the table of elements (Fe2+, Co2+, Ni2+, Cu2+, and Zn2+) led to the corresponding metalloporphyrins (1). As revealed by spectrophotometric analysis, most of the reactions were successful and yielded the desired metalloporphyrins. However, in the case of 4-(dimethylamino)benzaldehyde, 4-(diethylamino)benzaldehyde, and (pentamethyl)benzaldehyde the corresponding porphyrins were not formed and in the case of 4-hydroxy-benzaldehyde the obtained porphyrin did not undergo metallation with Fe2+ and Zn2+ ions. Overall, a library containing 83 metalloporphyrins was obtained which was used for automatic sequential screening of the catalytic activity of its members towards NO oxidation.
Fig. 1 shows photographs of the electrochemical robotic device that was used for automatically evaluating the properties of individual members of the porphyrin library with the focus on the positioning tables, the covered 96-well microtitre plate, and the easily positionable three-electrode assembly just above one of the wells. In general, differential pulse voltammetry was used to determine the catalytic efficiency of the metalloporphyrins at different concentrations of NO. At the beginning of a standardized automated test trial with a loaded microtitre plate, the electrochemical response of the freshly polished bare Pt disk working electrode against NO was measured in well D1 by serial additions of four 50 µL aliquots of NO stock solution to PBS containing 10 mM ferrocyanide as internal potential standard and recording the corresponding voltammograms (control; not shown). Next, multiple immersions into a well with water (F1) followed by potential sweeps in sulfuric acid in a potential range from −300 to +1200 mV (G1), and again dipping in water (H1) allowed for effective electrode cleaning. Good quality of the active surface of the Pt disk was confirmed by recording another voltammogram in the ferrocyanide–PBS solution in well D2 (next row). At this point, the sequence was continued in row 2 for screening the first metalloporphyrin (see Fig. 2). Residual water was removed from the working electrode by immersing the electrode assembly several times into dry DMF (A2) before repetitive dip-coating (3 times for 20 s) in the metalloporphyrin–DMF solution to modify the Pt disk with a thin layer of the given metalloporphyrin (B2). Freshly modified electrodes were allowed to stay in the Ar atmosphere with the electrode assembly positioned above the well for 5 minutes, and afterward were dipped into PBS in which they were cycled between 0 and 1000 mV to remove excess metalloporphyrin (C2). The washed porphyrin-modified Pt electrode was then subjected to serial additions of four 50 µL aliquots of NO stock solution and the related DPVs were recorded (D2). At the end of the cycle, the electrode was cleaned again in DMF (E2), water (F2), sulfuric acid under potential cycling between O2 and H2 evolution (G2), and finally water (H2). As with the metalloporphyrin in well B2, the programmed procedure was subsequently applied to the metalloporphyrins located in the wells B3 to B12.
 | ||
| Fig. 1 Photographs of the electrochemical robotic device that was used in conjunction with microtitre plates for automatically immobilizing metalloporphyrins on the surface of Pt disk electrodes and evaluating the properties of the resulting NO-sensitive sensors by means of in-well differential pulse voltammetry. Visible are the positioning tables, the covered 96-well microtitre plate, and the three-electrode assembly that is positioned just above one of the wells. | ||
 | ||
| Fig. 2 Typical standardized sequence of steps performed within an automated test trial for fabricating and screening porphyrin-based NO sensors in a charged microtitre plate. | ||
The ability to screen up to 10 metalloporphyrins per 96-well microtitre plate made 9 times repetition of the automated test trial necessary to evaluate the entire library of 83 synthesized metalloporphyrins and reveal their electrocatalytic properties through the analysis of the obtained raw data. The many differential pulse voltammograms within the datasets were displayed and determination of the corresponding peak potentials and peak currents as a function of well number (equal to a known metalloporphyrin) was performed taking advantage of a macro written in Origin® 6.1G (OriginLab Northampton, MA, USA). The reproducibility of the automatic sensor preparation and screening was tested by charging all the B2–B12 wells of one microtitre plate with the purchased MT-type porphyrin (Ni(II)tetrakis(3-methoxy-4-hydroxyphenyl)porphyrin), and subjecting the 11 in-well fabricated NO sensors to 4 sequential standard additions of NO stock solution. From these standardization measurements with the well-characterized commercial porphyrin, the standard deviations of the DPV peak potentials and currents for the programmed in-well sensor tests were determined to be about ±10 mV and ±11%, respectively.
Fig. 3 shows as a representative example a set of four DPVs obtained at a Pt electrode that was modified, in this case, with Ni(II)tetrakis(4-sulfomethylphenyl)porphyrin and effectively detected additions of four 50 µL aliquots of the NO stock solution through the oxidation of the analyte. For most of the screened porphyrins, well-formed peaks were observed with the peak values linearly depending on the actual concentration of NO in the PBS solution. In addition, the majority of the metalloporphyrin-modified electrodes demonstrated, more or less pronounced, the anticipated electrocatalytic effect for the oxidation of dissolved NO. In the case of a Pt disk, for example, that was dip-coated with Zn(II)tetrakis(4-bromophenyl)porphyrin, a nearly 5-fold increase in the peak current was obtained as compared with the response of the bare Pt electrode at the same NO concentration (see Fig. 4).
 | ||
| Fig. 3 A representative set of four differential pulse voltammograms obtained at a Pt electrode that was modified with Ni(II)tetrakis(4-sulfomethylphenyl)porphyrin. The sequential additions of four 50 µL aliquots of NO stock solution were effectively detected by in-well DPV. All DPVs were recorded in phosphate buffer solution using a pulse height of 150 mV and a pulse time of 60 ms. | ||
 | ||
| Fig. 4 Electrocatalytic effect of Zn/Br-porphyrin deposited on the surface of a Pt disk electrode. The DPV's of the bare Pt electrode (black trace) and modified NO sensor (grey trace) were recorded in phosphate buffer solution using a pulse height of 150 mV and a pulse time of 60 ms. | ||
The bar chart in Fig. 5 displays the current responses of all NO sensors based on the 16 synthesized Ni-porphyrins and weights their electrocatalytic power against a sensor that was prepared in the same way, however, using the commercial MT-type porphyrin as electrode modifier. Nine of the Ni-porphyrins synthesized within this trial served as superior, and another two as almost equal enhancers for the electrochemically induced oxidation of NO. An additional interesting observation was made when the total Hammett electronic parameters51 of the substituents on the meso-phenyl functionality of the porphyrins were calculated from individual values as listed elsewhere52 and included in Fig. 5 as scalar descriptors reflecting electron-donating or accepting properties. As is clearly visible, most of the 16 Ni-porphyrins that were at least as good as or better than the MT-type porphyrin carried substituents at the phenyl groups in the meso-positions of the porphyrin ring system that led to an overall Hammett constant of 0 or above. Apparently, the electron-withdrawing or -donating nature of the functional groups attached to the meso-phenyl groups appeared to have a considerable influence on the electrocatalytic activity of the associated porphyrins. This is consistent with earlier findings that made use of the Hammett methodology to get an insight into the factors influencing the catalytic activity of phthalocyanines and porphyrins.53–57
 | ||
| Fig. 5 Bar chart displaying the NO current responses of 16 Ni-porphyrins as compared with the catalytic current response of the comercially available (Malinski–Taha type) Ni-porphyrin. The numbers represent the total Hammett constants51 of the phenyl substituents in the meso-position of the porphyrin ring. | ||
A synopsis of the outcome from automatic screening of the catalytic properties of the metalloporphyrin library with the proposed robotic approach is provided in Table 1, which lists the NO peak currents and potentials determined for the individual members as far as they were accessible by means of automated differential pulse voltammetry. The peak potential for the electrocatalytic oxidation of NO at the individual porphyrin-modified Pt electrodes as measured vs. the oxidation of the internal standard varied for the metalloporphyrins in the library between about 449 (lowest, Ni/Ph) and 587 mV (highest, Ni/CF3). However, a low/high oxidation potential did not directly correlate with a good or bad catalytic activity of the related sensor. Furthermore, no correlation between a low/high NO oxidation potential and the sum of the Hammett constants of the substituents on the meso-phenyl rings was observable. The bar chart in Fig. 6 draws attention to the metalloporphyrin systems within the collection that offered catalytic current responses at least as good as the MT-type porphyrin. Actually, this was true for 37 (about 45%) of the 83 compounds of the library. As already demonstrated above for the subgroup of Ni porphyrins, most of the promising candidates had substituents on the meso-phenyl groups with overall Hammett-constant of 0 or above (see Fig. 7A).
 | ||
| Fig. 6 Bar chart listing all members of the metalloporphyrin library that showed catalytic current responses for the oxidation of NO at least as good as the commercially available (Malinski–Taha type) porphyrin. | ||
 | ||
| Fig. 7 (A) Circle diagram pooling the compounds with a positive (light grey), negative (dark grey) or zero (white) Hammett constant for the substituents on the meso-phenyl rings of the porphyrin macrocycle, calculated for a total of 37 porphyrin systems. The characteristic Hammett constants of the substituents were taken from literature (Hansch et al.52). The number of hits for the different substituents is also given. (B) Circle diagram illustrating the percentage of the different central metals (Fe, Zn, Ni, Cu, Co) from the total of 37 metalloporphyrins with a catalytic current response for the oxidation of NO at least as good as the commercially available (Malinski–Taha type) porphyrin. | ||
| R | Fe | Co | Ni | Cu | Zn |
|---|---|---|---|---|---|
| H | 538/2.55 | 529/1.59 | 534/4.36 | 531/2.43 | |
| Me | 525/0.77 | 528/1.98 | 520/0.71 | 500/0.83 | |
| i Pr | 509/0.85 | 541/1.38 | 556/1.58 | 465/1.39 | 489/1.29 |
| 2,4,6-Me | 516/2.43 | 522/1.50 | |||
| 2,3,4,5,6-Me | |||||
| OH | 527/1.58 | ||||
| OMe | 531/0.87 | 561/0.84 | 462/1.52 | 517/2.50 | |
| OEt | 524/1.05 | 525/0.70 | 534/0.88 | 488/1.29 | 500/0.85 |
| OBu | 487/014 | 502/1.74 | 547/0.92 | 536/1.39 | |
| Ph | 505/0.62 | 515/0.49 | 449/0.19 | 503/1.11 | 519/0.90 |
| NMe2 | |||||
| NEt2 | |||||
| SMe | 522/4.70 | 548/6.04 | 553/4.34 | 462/1.92 | 541/4.47 |
| CF3 | 580/5.93 | 579/3.30 | 587/3.43 | 581/5.60 | 579/3.19 |
| OCF3 | 517/1.61 | 509/0.27 | 561/3.46 | 537/1.84 | 494/2.39 |
| CN | 529/1.42 | 534/4.57 | 523/4.25 | 521/0.86 | 483/1.92 |
| NO2 | 546/1.97 | 532/2.36 | 520/12.8 | 528/3.94 | 541/1.29 |
| F | 495/0.50 | 558/1.54 | 498/1.99 | 497/0.62 | 520/2.66 |
| Cl | 501/1.97 | 520/2.56 | 454/1.55 | 472/1.98 | |
| Br | 509/2.86 | 525/2.47 | 497/3.04 | 529/4.44 | |
| M–T | 508/1.80 |
The metalloporphyrins with Zn/OMe, Co/Me and Ni/OH are the rare examples of exceptions featuring negative Hammett constants along with the required degree of electrocatalytic activity. Small differences in the polarity and/or hydrophobicity of the porphyrin side chains may also have an influence on the current response of the metalloporphyrin-modified Pt disk electrodes through modest variations in surface coverage, which obstructs a quantitative application of the Hammett methodology. On the other hand, the high-speed screening strategy with an in-well preparation of the NO sensors that is directly followed by an in-well characterization does not allow measurement of the actual surface coverage. However, from a qualitative point of view, the probability of getting effective metalloporphyrin-based NO microsensors is appreciably higher when choosing substituents on the meso-phenyl rings of the porphyrin macrocycle exhibiting overall positive Hammett constants. As further depicted in Fig. 7B, the five transition metals (Co, Cu, Fe, Ni, and Zn) that were utilised in the metalloporphyrin library investigated were represented almost equally in this collection with only Fe providing slightly fewer hits (see Fig. 7B).
In conclusion, an automated electrochemical approach for fabricating and testing metalloporphyrin-based electrocatalysts for NO sensors has been presented. A library of 83 metalloporphyrins with varying substitution pattern at the meso-position of the porphyrin ring and with different central metal ions has been examined using a specially designed electrochemical robotic system that allowed rather fast electrochemical measurements in microtitre-plate format. The technique was shown to be highly suitable for studying the effect of varied functionalities on the electrocatalytic properties of metalloporphyrins as catalytically active electrode modifiers that were deposited on Pt surfaces by means of automatic dip-coating. Structure-based catalyst design in combination with computerized catalyst immobilization and screening decreased the effort associated with sensor screening and helped with the identification of good and elimination of poor catalyst candidates. A definite plus of automatically performing the immobilization of the porphyrin films and subjecting them afterward to programmed differential pulse voltammetry is the high degree of repeatability and reproducibility due to the exclusion of manual errors. Screening the 83 members of the porphyrin library for their performance with respect to the electrooxidation of NO made it obvious that there is a clear link between the calculated value of the Hammett constant of the functional groups attached to the meso-phenyl groups and the achievable electrocatalytic activity of the corresponding metalloporphyrin layer. Further optimization, even of already well-established systems such as porphyrin-based NO sensors, thus may well be achievable via an optimal choice of the chemical composition of the electrocatalyst; therefore a predictive and more rational approach to the design of chemically modified sensors should be targeted.
Acknowledgements
Part of the work was financially supported initially by the DFG (Schu 929/4-1) in the framework of the co-operative project “Kombinatorische Mikroelektrochemie” and the Ministry of Education and Research, Germany (BMBF) in the framework of its program “Nanobiotechnologie” (AZ NBT066). The authors are grateful to Armin Lindner and his colleagues from the mechanical workshop of the faculty of chemistry at the Ruhr-Universität Bochum.References
- J. Lincoln, C. H. V. Hoyle and G. Burnstock, Nitric oxide in health and disease, Cambridge University Press, Cambridge, UK, 1997 Search PubMed.
- Z. H. Taha, Talanta, 2003, 61, 3–10 CrossRef CAS.
- L. Huang and R. T. Kennedy, Trends Anal. Chem., 1995, 14, 158–164 CrossRef CAS.
- M. Pontié and F. Bedioui, Analusis, 2000, 28, 465–469 CrossRef CAS.
- F. Bedioui and N. Villeneuve, Electroanalysis, 2003, 15, 5–17 CrossRef CAS.
- R. D. Hurst and J. B. Clark, Sensors, 2003, 3, 321–329 Search PubMed.
- X. Zhang, Front. Biosci., 2004, 9, 3434–3446 Search PubMed.
- T. Malinski and Z. Taha, Nature, 1992, 358, 676–678 CrossRef CAS.
- T. Malinski, Z. Taha, S. Grunfeld, A. Burewicz, M. Tomboulian and F. Kiechle, Anal. Chim. Acta, 1993, 279, 135–140 CrossRef CAS.
- S. Trevin, F. Bedioui, M. G. Gomez Villagas and C. Bied-Charreton, J. Mater. Chem., 1997, 7, 923–928 RSC.
- J. Y. Chen, O. Ikeda, T. Hatasa, A. Kitajima, M. Miyake and A. Yamatodami, Electrochem. Commun., 1999, 1, 274–277 CrossRef CAS.
- M. Biesaga, K. Pyrzynska and M. Trojanoviz, Talanta, 2000, 51, 209–224 CrossRef CAS.
- N. Diab and W. Schuhmann, Electrochim. Acta, 2001, 47, 265–273 CrossRef CAS.
- N. Diab, J. Oni, A. Schulte, I. Radtke, A. Blöchl and W. Schuhmann, Talanta, 2003, 61, 43–51 CrossRef CAS.
- L. Mao, K. Yamamoto, W. Zhou and L. Jin, Electroanalysis, 2000, 12, 72–77 CrossRef CAS.
- T. Nyokong and S. L. Vilakazi, Talanta, 2003, 61, 27–35 CrossRef CAS.
- A. Pailleret, J. Oni, S. Reiter, S. Isik, M. Etienne, F. Bedioui and W. Schuhmann, Electrochem. Commun., 2003, 5, 847–852 CrossRef CAS.
- P. M. Doyle, J. Chem. Technol. Biotechnol., 1995, 64, 317–324 CrossRef CAS.
- X. Willard, I. Pop, L. Bourel, D. Horvath, R. Baudelle, P. Melnyk, B. Deprez and A. Tartar, Eur. J. Med. Chem., 1996, 31, 87–98 CrossRef.
- L. M. Amzel, Curr. Opin. Biotechnol., 1998, 9, 366–369 CrossRef CAS.
- P. W. Erhardt, Pure Appl. Chem., 2002, 74, 703–785 CrossRef CAS.
- O. Schwardt, H. Kolb and B. Ernst, Curr. Top. Med. Chem., 2003, 3, 1–9 Search PubMed.
- O. W. Gooding, Curr. Opin. Chem. Biol., 2004, 8, 297–304 CrossRef CAS.
- J. C. Reader, Curr. Top. Med. Chem., 2004, 4, 671–686 Search PubMed.
- F. Gennari, P. Seneci and S. Miertus, Catal. Rev. Sci. Eng., 2000, 42, 385–402 CrossRef CAS.
- S. Senkan, Angew.Chem., Int. Ed., 2001, 40, 312–329 CrossRef CAS.
- H. Wennemers, Comb. Chem. High Throughput Screening, 2001, 4, 273–285 Search PubMed.
- A. Hagemeyer, B. Jandeleit, Y. M. Liu, D. M. Poojary, H. W. Turner, A. F. Volpe and W. H. Weinberg, Appl. Catal., A, 2001, 221, 23–43 CrossRef CAS.
- T. Siu, S. Yekta and A. K. Yudin, J. Am. Chem. Soc., 2000, 122, 11787–11790 CrossRef CAS.
- C. A. Briehn and P. Bäuerle, J. Comb. Chem., 2002, 4, 457–469 CrossRef CAS.
- C. Barbero, H. J. Salavagione, D. F. Acevedo, D. E. Grumelli, F. Garay, G. A. Planes, G. M. Morales and M. C. Miras, Electrochim. Acta, 2004, 49, 3671–3686 CrossRef CAS.
- V. Kulikov and V. M. Mirsky, Meas. Sci. Technol., 2004, 15, 49–54 CrossRef CAS.
- G. Gilardi, A. Fantuzzi and S. J. Sadeghi, Curr. Opin. Struct. Biol., 2001, 11, 491–499 CrossRef CAS.
- S. Reiter, K. Eckhard, A. Blöchl and W. Schuhmann, Analyst, 2001, 126, 1912 RSC.
- M. G. Sullivan, H. Utomo, P. J. Fagan and M. D. Ward, Anal. Chem., 1999, 71, 4369–4375 CrossRef CAS.
- W. A. Steen and D. T. Schwartz, Chem. Mater., 2003, 15, 2449–2453 CrossRef CAS.
- M. Devenney, S. W. Donne and S. Gorer, J. Appl. Electrochem., 2004, 34, 643–651 CrossRef CAS.
- M. D. Fleischauer, T. D. Hatchard, G. P. Rockwell, J. M. Topple, S. Trussler, S. K. Jericho, M. H. Jericho and J. R. Dahn, J. Electrochem. Soc., 2003, 150, A1465–A1469 CrossRef CAS.
- K. Takada, K. Fujimoto, T. Sasaki and M. Watanabe, Appl. Surf. Sci., 2004, 223, 210–213 CrossRef CAS.
- E. Reddington, A. Sapienza, B. Gurau, R. Viswanathan, S. Sarangapani, E. S. Smotkin and T. E. Mallouk, Science, 1998, 280, 1735–1737 CrossRef CAS.
- R. Z. Jiang and D. Chu, J. Electroanal. Chem., 2002, 527, 137–142 CrossRef CAS.
- E. S. Smotkin and R. R. Diaz-Morales, Annu. Rev. Mater. Res., 2003, 33, 557–579 Search PubMed.
- S. Guerin, B. E. Hayden, C. E. Lee, C. Mormiche, J. R. Owen, A. E. Russel, B. Theobald and D. Thompsett, J. Comb. Chem., 2004, 6, 149–158 CrossRef CAS.
- S. Jayaraman and A. C. Hillier, J. Comb. Chem., 2004, 6, 27–31 CrossRef CAS.
- B. Ngounou, S. Neugebauer, A. Frodl, S. Reiter and W. Schuhmann, Electrochim. Acta, 2004, 49, 3855–3863 CrossRef CAS.
- S. Reiter, D. Ruhlig, B. Ngounou, S. Neugebauer, S. Janiak, A. Vilkanauskyte, T. Erichsen and W. Schuhmann, Macromol. Rapid Commun., 2004, 25, 348–354 CrossRef CAS.
- Y. P. Sun, H. Buck and T. E. Mallouk, Anal. Chem., 2001, 73, 1599–1604 CrossRef CAS.
- T. Erichsen, S. Reiter, W. Märkle, C. Tittel, V. Ryabova, E. M. Bonsen, G. Jung, B. Speiser and W. Schuhmann, Rev. Sci. Instrum., in press Search PubMed.
- C. A. Briehn, M.-S. Schiedel, E. M. Bonsen, W. Schuhmann and P. Bäuerle, Angew. Chem., Int. Ed., 2001, 113, 4817–4820 CrossRef.
- F. Li, K. Yang, J. S. Tyhonas, K. A. MacCrum and J. A. Lindsey, Tetrahedron, 1997, 53, 12339–12360 CrossRef CAS.
- L. P. Hammett, Physical Organic Chemistry, McGraw Hill, New York, 1970 Search PubMed.
- C. Hansch, A. Leo and R. W. Taft, Chem. Rev., 1991, 91, 165–195 CrossRef CAS.
- M. Yuasa, B. Steiger and F. C. Anson, J. Porphyrins Phthalocyanines, 1997, 1, 181–188 CrossRef CAS.
- A. B. P. Lever, J. Porphyrins Phthalocyanines, 1999, 3, 488–499 CrossRef CAS.
- T. V. Magdesieva, I. V. Zhukov, L. G. Tomilova, O. V. Korenchenko, I. P. Kalashnikova and K. P. Butin, Russ. Chem. Bull., 2001, 50, 396–403 CrossRef CAS.
- M. J. Aguirre, M. Isaacs, F. Armijo, L. Basáez and J. H. Zagal, Electroanalysis, 2002, 14, 356–362 CrossRef CAS.
- C. Linares, D. Geraldo, M. Paez and J. H. Zagal, J. Solid State Electrochem., 2003, 7, 626–631 Search PubMed.
| This journal is © The Royal Society of Chemistry 2005 |