Screening of specific antigens for SARS clinical diagnosis using a protein microarray
Dan-Dan
Lu
,
Su-Hong
Chen
,
Shi-Meng
Zhang
,
Min-Li
Zhang
,
Wei
Zhang
,
Xiao-Chen
Bo
and
Sheng-Qi
Wang
*
Beijing Institute of Radiation Medicine, Beijing, 100850, People's Republic of China. E-mail: sqwang@nic.bmi.ac.cn; Fax: +86-10-68214653; Tel: +86-10-66932211 and +86-10-66931422
First published on 14th February 2005
Abstract
In this study several SARS-CoV structural proteins and fragments were expressed in E. coli as GST or TRX fusion proteins. They were fabricated on a microarray and tested with sera from SARS patients. Antigenic screening indicated that recombinant GST-N2 fusion protein, the carboxy-terminus 213aa–423aa of N protein, was strongest positive and weakest non-specific compared with others. An indirect antibody ELISA method was developed and clinical positive and negative sera for their antibodies against GST-N2 fusion protein were assayed. 311 out of the 442 sera from clinical SARS inpatients, as well as 229 out of 302 sera from convalescent patients gave positive reactivities; positive rates were 70.4% and 75.8% respectively. Sera from a total of 2726 non-SARS patients and healthy individuals were tested and the false positive rate was only 0.07%. When the sensitivity control sample was diluted 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) ∶64, it yielded OD values above the cutoff value. Reported data showed that this was a relatively high degree of sensitivity and specificity for SARS-CoV antibody testing. The data indicate that GST-N2 fusion protein, which was screened by protein microarray, may be a valuable diagnostic antigen for the development of serological assays for SARS. In addition, protein microarray assay presents a higher positive rate and sensitivity (86.1% and 1∶200) compared with the traditional ELISA screening method, and could provide a rapid, parallel and high-throughput antigen screening platform.
∶64, it yielded OD values above the cutoff value. Reported data showed that this was a relatively high degree of sensitivity and specificity for SARS-CoV antibody testing. The data indicate that GST-N2 fusion protein, which was screened by protein microarray, may be a valuable diagnostic antigen for the development of serological assays for SARS. In addition, protein microarray assay presents a higher positive rate and sensitivity (86.1% and 1∶200) compared with the traditional ELISA screening method, and could provide a rapid, parallel and high-throughput antigen screening platform.
Introduction
The global outbreak of severe acute respiratory syndrome (SARS) in 2003 greatly threatened public health. It is recognized that SARS-associated coronavirus (SARS-CoV) was primarily responsible for SARS infection, designated a novel coronavirus by the World Health Organization (WHO).1–4 Within a short period, SARS-CoV was decoded completely, providing a fundamental basis for understanding its pathogenesis and developing effective diagnostic and therapeutic approaches.5–7Bioinformatics analysis indicates that SARS-CoV is an enveloped, single-stranded, positive-sense RNA virus classified within the Coronaviridae family. The genome of SARS-CoV is approximately 29727 nucleotides, which has 11 open reading frames, encoding four structural proteins: spike (S) protein, membrane (M) protein, nucleocapsid (N) protein and the small envelope (E) protein.5
Antigenicity studies with other coronaviruses indicated that the S protein is the main surface antigen. Enjuanes et al. also reported S protein as the primary neutralization antigen of coronaviruses.8 It is composed of a globular domain at the amino-terminus and a transmembrane claviform structure at the carboxy-terminus. S protein takes part in viral adhesion to the host cell and infection courses.9 M protein plays an important role in receptor binding and virus budding. It is necessary that M protein interacts with S protein to combine the virus membrane.10 With a length of only 72 amino acids, E protein has been taken as an indispensable membrane component of coronaviruses. N protein is one of the immunodominant antigens that induce cross-reactive antibodies in high titers. In addition, N protein has multiple functions that are involved in providing nuclear-import signal, interfering in cell processes, virus replication and RNA packaging.
Although the SARS outbreak has ended, people still have quite limited knowledge about this infectious disease; in particular there has not been an efficient clinical diagnosis method. An accurate diagnosis method for SARS is urgently needed and it will be a great help for rapid, sensitive clinical diagnosis of SARS and prevention of SARS recurrence. Protein microarray has developed as a powerful tool to study gene expression and antibody screening in recent years.11 This assay has key features, such as high-throughput, miniaturization and true parallelism.12 Protein microarrays were initially employed as diagnostic immunoarrays, in which several sample parameters could be simultaneously analyzed. It has been demonstrated that a protein microarray assay can be employed to determine in human sera the presence or absence of specific antibodies directed against a variety of antigens.13 In this study, a protein microarray has been generated for high-throughput detection and screening of recombinant SARS-CoV antigens. It aimed to find specific antigen fragments that are of use in developing a clinical SARS antibodies testing method, such as ELISA (enzyme-linked immunosorbent assay).
Materials and methods
Bacteria
For cloning and expression of the SARS-CoV fusion proteins and fragments, plasmids harboring the corresponding chimeric genes were transformed into Escherichia coli (E. coli) strains DH5α and BL21 (DE3).Sera
Sera from individuals were tested for anti-SARS proteins reactivity. They were obtained from 744 clinically diagnosed SARS patients, 50 probable SARS patients, 1566 non-SARS patients and 1160 healthy individuals supplied by four hospitals, from March to May, 2003. Control sera included 10 positive sera, 20 negative sera and 1 serially diluted standard positive sera as sensitivity control, supplied by the National Institute for the Control of Pharmaceutical and Biological Products.Construction of plasmids
The nucleotide sequences coding for the entire SARS N gene, two partials of M gene fragments and four partials of S gene fragments were obtained by the polymerase chain reaction (PCR) using the entire SARS-CoV genome cDNA. M gene contains 666 bp nucleotides (nt 26383–27048), N gene contains 1269 bp nucleotides (nt 28105–29373), S gene contains 3768 bp nucleotides (nt 21477–25244). Full-length N gene, two M gene fragments M1, M2 (1–333bp, 334–666bp) and four S gene fragments S1, S2, S3, S4 (1–924 bp, 925–1848 bp, 1900–2823 bp, 2824–3768 bp) were cloned into pGEX-4T-1 (Amersham Pharmacia Biotech), PET-28a (Novagen), or PET-32a (Novagen) vectors. Oligonucleotide primers were deduced from the genomic sequences of respective genes (Table 1).
| Designation | Sequence (5′–3′) |
|---|---|
| Z3 | 5′-GC GGATCC ATG TCT GAT AAT GGA CCC CAA-3′ |
| Z4 | 5′-CC GAATTCTTA CTT TGC CTG AGT TGA ATC-3′ |
| M1F | 5′-GC GGATCC ATG GCA GAC AAC GGT ACT ATT-3′ |
| M1R | 5′-CC GAATTC GAA TGA CCA CAT TGA GCG GGT-3′ |
| M2F | 5′-GC GGATCC AAC CCA GAA ACA AAC ATT CTT-3′ |
| M2R | 5′-CC GAATTCTTA CTT CTG TAC TAG CAA CGC-3′ |
| S1F | 5′-GC GGATCC ATG TTT ATT TTC TTA TTA TTT-3′ |
| S1R | 5′-CC GAATTC AAC AAC CCT GAA ATT AGA GGT-3′ |
| S2F | 5′-GC GGATCC CCC TCA GGA GAT GTT GTG AGA-3′ |
| S2R | 5′-CC GAATTC TAA TTT TTG CTT GAG TCT GGA ATA-3′ |
| S3F | 5′-GC GGATCC GGC TGT CTT ATA GGA GCT GAG-3′ |
| S3R | 5′-CC GAATTC TAA TGC TTG AGC ATT CTG GTT-3′ |
| S4F | 5′-GC GGATCC AAC ACA CTT GTT AAA CAA CTT-3′ |
| S4R | 5′-CC GAATTCTTA CTT TGT GTA ATG TAA TTT GAC-3′ |
In order to further identify the antigenic fragments of the N protein and to screen the specific antigens for the SARS diagnosis, nine parts were divided according to the sequence of N protein. We designed and synthesized the primers according to the N gene sequence (Table 2).
| Designation | Sequence (5′–3′) |
|---|---|
| N1F | 5′-GC GGATCC ATG TCT GAT AAT GGA CCC CAA-3′ |
| N1R | 5′-CC GAATTC GTG ATG ATG ATG ATG ATG GGG GCC GTC ACC ACC ACG AAC-3′ |
| N2F | 5′-GC GGATCC ATG AAA GAG CTC AGC CCC AGA-3′ |
| N2R | 5′-CC GAATTC GTG ATG ATG ATG ATG ATG AGC CAT TCG AGC AGG AGA ATT-3′ |
| N3F | 5′-GC GGATCC AGC GGA GGT GGT GAA ACT GCC-3′ |
| N3R | 5′-CC GAATTC GTG ATG ATG ATG ATG ATG GAA TTC TCC AAA GAA TGC AGA-3′ |
| N4F | 5′-GC GGATCC ATG GAA GTC ACA CCT TCG GGA-3′ |
| N4R | 5′-CC GAATTC GTG ATG ATG ATG ATG ATG CTT TGC CTG AGT TGA ATC-3′ |
The sense primers contained BamH I restriction sites (italicized), while the antisense primers contained EcoR I restriction sites (italicized) and some contained a stop codon (bold). In the antisense primers of N gene fragments, we designed six codons of histidine to construct double tag (GST-tag and His-tag) vectors. The amplified genes were treated with BamH I and EcoR I restriction enzymes (TaKaRa), blunt ended and ligated into the corresponding sites of the pGEX-4T-1, PET-28a or PET-32a express vectors with T4 ligase (TaKaRa). Constructed expression plasmids were transformed into E. coli DH5α and BL21 (DE3) host cells. The N gene nucleotide sequence was cloned into the PET-28a vector (P28-N). Nine partials of N gene fragments and E gene nucleotide sequence were cloned into the pGEX-4T-1 vector (4T-1-CN1–CN7, 4T-1-N1, 4T-1-N2), resulting in plasmids that contained the N-terminus of the N and E nucleotide sequences that fused to the glutathione S-transferase (GST) gene. Two fragments of M gene nucleotide sequence and four fragments of S gene nucleotide sequence were cloned into the PET-32a vectors (P32-M1, P32-M2, P32-S1, P32-S2, P32-S3, P32-S4), resulting in plasmids that contained the N-terminus of the M and S nucleotide sequences that fused to the thioredoxin protein (TRX) gene. pGEX-4T-1 and PET-32a empty vectors were transformed into E. coli DH5α and BL21 (DE3) host cells as control. Cloning was performed according to standard protocols.14
Expression and purification of SARS recombinant proteins
The above constructed plasmids were transformed into the E. coli strain DH5α (PET-28a series plasmids required addition of kanamycin and pGEX-4T-1 and PET-32a series plasmids required addition of ampicillin). To isolate plasmids, a single positive colony was selected and transformed into E. coli strain BL21 (DE3). The plasmids were induced to synthesize the fusion proteins by the addition of isopropyl-β-D-thiogalactopyranoside (IPTG) (Merck). Pilot experiments were performed in order to find the optimized conditions for protein expression. It was found that maximum expression of the proteins occurred when 4T-1-CN1–CN7, 4T-1-N1, 4T-1-N2 and P28-N were induced with 0.5 mM IPTG for 4 h at 37 °C and when P32-M1, P32-M2, P32-S1, P32-S2, P32-S3 and P32-S4 were induced with 0.1 mM IPTG for 3 h at 25 °C. Cells were collected by centrifugation and the pellet suspended by addition of PBS (137 mM NaCl, 2.7 mM KCl, 10 mM Na2HPO4, 2 mM KH2PO4, pH 7.4). The suspension was centrifuged at 5000 × g at 4 °C for 5 min and the supernatant was discarded. The harvested cells expressing the GST fusion proteins were suspended in sonication buffer (PBS, 1 mM DDT, 1 mM PMSF) and cells expressing proteins containing the His-tag were suspended in sonication buffer [0.02 M PB (0.02 M Na2HPO4, 0.02 mM NaH2PO4, pH 7.2), 0.5 M NaCl, pH 8.0, 1 mM DDT, 1 mM PMSF]. Sonication was performed in an ice bath. The pellet was separated by centrifugation at 10000 ×
g at 4 °C for 10 min. Proteins with N-terminus fused to a His-tag were purified from the supernatant solution by a Ni2+ affinity column (Amersham Pharmacia Biotech) and proteins with N-terminus fused to GST by a Glutathione Sepharose 4B column (Amersham Pharmacia Biotech) as suggested by manufacturer's instruction. The purified fusion proteins were separated on 12% SDS-PAGE and stained with Coomassie blue. PET-32a vector protein TRX and pGEX-4T-1 vector protein GST were expressed in E. coli and purified by Ni2+ affinity column and GST affinity column. The molecular weight of purified GST-N2 fusion protein was confirmed by MALDI-TOF-MS (REFLEXTM III Bruker Co.).
Preparation of the protein microarrays
Protein microarrays were prepared for the detection of anti-SARS antibodies in human sera and for screening specific antigens for the development of clinical diagnosis. The purified SARS-CoV recombinant proteins, TRX protein and GST protein were dissolved in printing buffer (0.02% SDS and 1% glycerol respectively) and direct contact-printed15,16 on aldehyde-activated glass slides, using a computer-controlled microchip spotting instrument (Cartisan Pixsys 3000). Samples were transferred from 384 microtiter plates to glass slides by using stainless steel micro spotting pins (400 µm diameter). Each pin was estimated to transfer ∼2 nL of sample to the slide. The size of glass slide was 25.4 mm × 76.2 mm, including 10 matrixes (7.5 mm × 7.5 mm). Two kinds of screening microarrays were prepared, consisting of various SARS-CoV structure proteins (total 45 slides) and N protein fragments (total 15 slides). Cross-reactions were tested using microarrays printed with purified GST and TRX proteins. Each antigen was printed in three replicates within a matrix. Decreasing amounts of human IgG (200 pg, 100 pg, 50 pg, 20 pg, 4 pg and 2 pg, Sigma) were used as positive control and purified GST protein as negative control in N fragment screening microarrays (Fig. 1 A). SARS-CoV structure protein screening microarrays were similar, but differed in the printed antigens and in that a fixed amount of human IgG (200 pg) was printed as coordinate (Fig. 1 B). The printing process was performed at 25 °C and 55% humidity.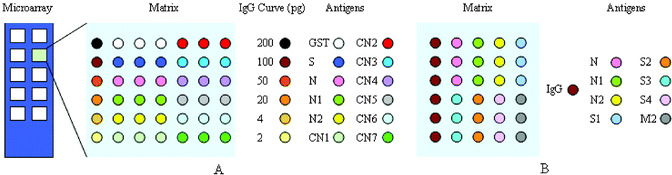 | ||
| Fig. 1 Schematic representation of protein microarrays used in this study. (A) N protein fragment screening microarray; (B) SARS-CoV structure protein screening microarray. Colored circles indicate printing positions of antigens and the left column within each matrix was human IgG coordinate. | ||
Processing of protein microarrays
Protein microarrays were blocked with PBS containing 25% foetal calf serum (FCS) overnight at 4 °C, then 37 °C for 1 h before removal of the blocking buffer. Printed slides can be used immediately or stored. Protein microarrays were washed with PBST (137 mM NaCl, 2.7 mM KCl, 10 mM Na2HPO4, 2 mM KH2PO4, 0.05% v/v Tween 20, pH 7.4) before processing. Serum samples were diluted 1∶10 in dilution buffer (PBS containing 10% FCS) and incubated at 37 °C for 30 min in a humid box. The slides were washed and subsequently incubated at 37 °C for 30 min with mouse anti-human IgG antibody labeled with Cy3 fluorophores (Amersham Pharmacia Biotech)
(1∶200 dilution). Before the fluorescence was read in the scanner, the slides were washed and dried at room temperature. The fluorescence intensity emitted by the fluorophore at 532 nm wavelength was detected by a GenePix 4000 scanner (Axon). Pseudo-color scale images were auto-generated and the fluorescence intensity of each spot on the microarrays was automatically analyzed using GenePix Pro 4.0 software (Axon). The SARS-CoV structure protein screening microarrays were used to qualitatively screen the antigenicity of various structure proteins by comparing the mean fluorescence intensity of 100 negative sera against GST and TRX proteins respectively plus 3 standard deviations (SD). N protein fragment screening microarrays contained internal calibration curves generated by the decreasing amounts of IgG printed. The amounts of IgG in sera were calculated by interpolating the fluorescence intensity of each spot into the IgG dose–response curves.
Establishment of recombinant SARS-CoV fusion antigen GST-N2 ELISA
Microtiter plates (Costar) were coated with 100 µl of the fusion antigens GST-N2 or the control GST, at a final concentration of 200 ng well−1 in 0.1 M carbonate buffer (pH 9.6). Unbound antigen was removed and the wells were blocked with 150 µl PBS supplemented with 5% FCS at 4 °C overnight. After blocking, the wells were washed five times with PBST. 50 µl serum was added to each well and the plate was incubated for 1 h at 37 °C. The plate was washed as described previously. Then 50 µl HRP-coupled rabbit anti-human IgG (Sigma) at 1∶1000 in PBST solution was added to each well and the plate was incubated for 1 h at 37 °C. Bound antibodies were detected by the addition of 100 µl TMB (3,3′,5,5′-tetramethylbenzidine) substrate solution and colorimetric determination of the absorbance at 450 nm by a microplate autoreader (Wallac). For each sample, the final absorbance value was calculated by subtracting the OD450 of the control antigen ELISA from the respective OD450 of GST-N2 fusion protein ELISA. The cutoff value was determined as the mean value of the 616 samples from the healthy individuals plus 3 SD.
Results
Expression of recombinant proteins of SARS-CoV in E. coli
To obtain efficient expression of SARS-CoV proteins in E. coli, three expression vectors, pGEX-4T-1 (Amersham Pharmacia Biotech), PET-28a (Novagen), and PET-32a (Novagen) were used to produce the recombinant SARS-CoV proteins and fragments. Seven expression plasmids were constructed. Two fragments of M protein and four fragments of S protein fused to TRX (TRX-M1, TRX-M2, TRX-S1, TRX-S2, TRX-S3 and TRX-S4) were expressed in E. coli in both soluble and inclusion body forms, while full-length N protein fused to His-tag was expressed in soluble form in E. coli. Subsequently, based on the antigenic screening results by protein microarray, we constructed nine plasmids containing the different length fragments of N gene sequence (Fig. 2). Nine partial fragments of N protein (GST-N1, GST-N2, GST-CN1–CN7) fused to GST. All the expression of the recombinant SARS-CoV proteins and peptides are summarized in Table 3. Total cell lysates from E. coli induced with IPTG were separated on 12% SDS-PAGE and the proteins and peptides were visualized by Coomassie blue staining (Figs. 3 and 4). TRX and GST were expressed and purified as control, shown in Fig. 5. The molecular weight of GST-N2 fusion protein was 48075 Daltons by MALDI-TOF-MS analysis, which was consistent with the electrophoresis result (Fig. 6).
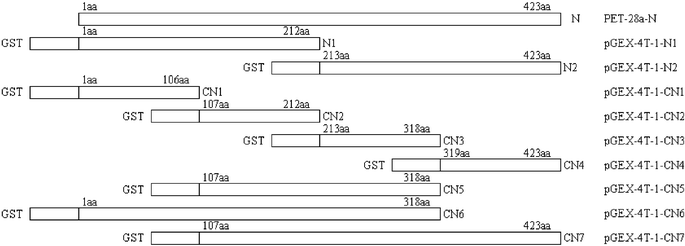 | ||
| Fig. 2 Schematic representation depicting the coding domain for nine partials of N protein fused to GST. Rectangles indicate the different amino acid sequences of full-length N protein and N protein fragments fused to the GST. Numbers indicate amino acid positions. | ||
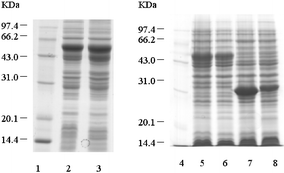 | ||
| Fig. 3 SDS-PAGE of the expressed recombinant SARS-CoV S and M protein fragments. Lane 1 and Lane 4: low molecular protein standards. Lane 2: P32a-S1 cell lysates. Lane 3: P32a-S2 cell lysates. Lane 5: P32a-S3 cell lysates. Lane 6: P32a-S4 cell lysates. Lane 7: P32a-M1 cell lysates. Lane 8: P32a-M2 cell lysates. | ||
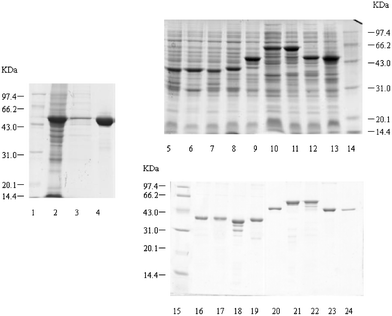 | ||
| Fig. 4 SDS-PAGE of expressed and purified recombinant SARS-CoV N protein and polypepides. Lane 1, Lane 14 and Lane 15: low molecular protein standards. Lane 2: P28-N cell lysates. Lane 3: P28-N cell supernatant. Lane 4: purified N protein. Lane 5 to Lane 11: 4T-1-CN1–CN7 cell lysates. Lane 12: 4T-1-N1 cell lysates. Lane 13: 4T-1-N2 cell lysates. Lane 16 to Lane 22: purified GST-CN1–CN7 proteins. Lane 23: purified GST-N1 protein. Lane 24: purified GST-N2 protein. | ||
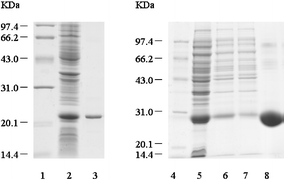 | ||
| Fig. 5 SDS-PAGE of expressed and purified TRX and GST. Lane 1 and Lane 4: low molecular protein standards. Lane 2: PET-32a cell lysates. Lane 3: purified TRX. Lane 5: pGEX-4T-1 cell lysates. Lane 6 and Lane 7: pGEX-4T-1 cell supernatant. Lane 8: purified GST. | ||
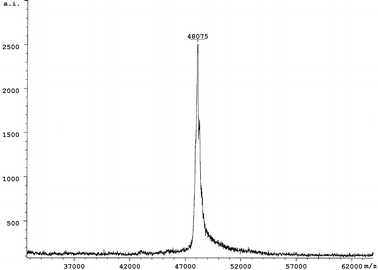 | ||
| Fig. 6 The molecular weight determination of GST-N2 protein using MALDI-TOF-MS. | ||
| Designation | Coding domain length/aa | Molecular weight/kDa | Designation | Coding domain length/aa | Molecular weight/kDa |
|---|---|---|---|---|---|
| TRX-M1 | 111 | 33 | GST-N1 | 212 | 49 |
| TRX-M2 | 111 | 31 | GST-N2 | 211 | 49 |
| TRX-S1 | 308 | 55 | GST-CN1 | 106 | 39 |
| TRX-S2 | 308 | 55 | GST-CN2 | 106 | 39 |
| TRX-S3 | 308 | 55 | GST-CN3 | 106 | 39 |
| TRX-S4 | 308 | 53 | GST-CN4 | 105 | 39 |
| N | 423 | 50 | GST-CN5 | 212 | 49 |
| TRX | 21 | GST-CN6 | 318 | 60 | |
| GST | 26 | GST-CN7 | 317 | 60 |
Preliminary screening of the antigenic specificity to recombinant SARS-CoV proteins and fragments by protein microarrays
Sera from 122 clinically diagnosed SARS patients, 50 probable SARS patients and 100 healthy individuals were processed by protein microarrays as described above. GST and TRX protein were printed on the microarray in order to ascertain whether they cross-reacted with the antibodies in the sera. Fig. 7 shows GST and TRX hybridization with a negative sample and a positive sample. Evidently, TRX presented non-specific reaction with negative sera, which could disturb the detection, however, GST did not. This suggested that the GST was more suitable to being fused to recombinant SARS-CoV proteins than TRX.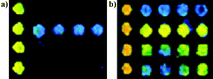 | ||
| Fig. 7 TRX and GST hybridization with (a) negative serum and (b) positive serum. The first row was GST and the second row was TRX. | ||
One hundred negative sera were tested against purified GST and TRX proteins and mean detection limits of the fluorescence intensity were 230 and 550 respectively. SARS-CoV structure protein screening microarrays were used to detect the reactivity of sera to various SARS-CoV proteins and fragments. The fragments of S protein fused to TRX showed strong false positive (averaging 70%) to the negative sera compared with the TRX mean detection limits. Furthermore, we could not distinguish whether the positive reactivity came from TRX or the S protein fragments themselves. No positive reactivity to the negative sera was determined for GST-N2 fusion protein, while 105 out of 122 sera from SARS patients showed positive reactivity (positive rate of 86.1%). Representative results of a typical protein microarray analysis for 5 positive sera and 5 negative sera are presented in Fig. 8.
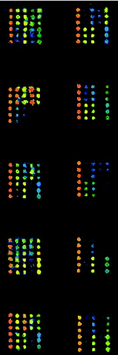 | ||
| Fig. 8 Scan images of SARS-CoV structure protein screening microarrays incubated with 5 positive sera and 5 negative sera at 532 nm detection wavelength. The left column is positive and the right column is negative. | ||
Analysis of IgG calibration curves and the antigenicity of GST-N2 protein fragments
Internal calibration curves were generated by printing about 2 nL of decreasing concentrations (100 µg ml−1, 50 µg ml−1, 25 µg ml−1, 10 µg ml−1, 2 µg ml−1, and 1 µg ml−1) of human IgG solution. The reactivities of the secondary antibodies against the negative control (GST protein) were used as the first point of the calibration curves. Regression analysis demonstrated that IgG dose–response curves had similar slopes (CV < 15%) and coefficients of relativity (r2) close to 1 (0.97–0.99 in these arrays). It was indicated that the internal calibration curves of IgG showed a high reproducibility and linear relationship under these experimental conditions (Figs. 9 and 10). The amounts of IgG in these sera against different antigen spots were determined by interpolating the fluorescence intensity into the internal calibration curves. These sera showed different reactivities against various N protein fragments (Figs. 9 and 10). It was obvious that a carboxy-terminal of N protein (N2) of about 200 aa in length showed the most strongly positive signals when probed with sera from SARS patients and had a negative reaction with sera from healthy individuals.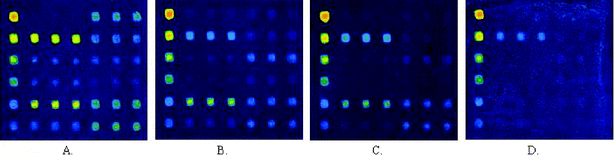 | ||
| Fig. 9 Scan images of N protein fragment screening microarrays incubated with strong positive (A), medium positive (B), weak positive (C), and negative (D) serum samples at 532 nm detection wavelength. | ||
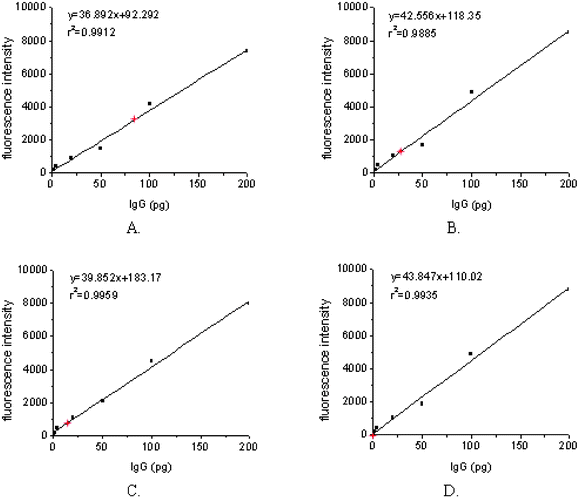 | ||
| Fig. 10 IgG amounts were calculated by interpolating mean fluorescence intensity collected from arrayed GST-N2 proteins, incubated with strong positive (A), medium positive (B), weak positive (C), and negative (D) serum samples into the IgG dose–response curves (red stars). | ||
The detection limit, defined as mean fluorescence intensity of the negative control plus 3SD interpolated into the IgG internal calibration curves, was 7.1 pg. The results on 20 negative control sera and 10 positive control sera showed that this assay had a high specificity (0/20) and good sensitivity (10/10). The reactivity against N2 protein was positive when the sensitivity control serum was diluted 1∶200. Comparing serum reactivity against GST-N2 protein of different IgG concentrations, the CV values were 1.3% to 11.6% and 4.7% to 18.5%, within and between slides respectively (n
= 3 and n
= 4).
Analysis by ELISA of clinical patient sera for antibodies against recombinant GST-N2 fusion protein
According to antigen screening by protein microarrays, recombinant GST-N2 protein has strong antigenicity in all SARS-CoV proteins. GST-N2 fusion protein was purified from bacterial lysates by GST affinity chromatography and assessed for the development of ELISA as described in Materials and Methods. The cutoff value of GST-N2 ELISA was 0.116 [0.050 ± 3 × 0.0333 (mean ± 3 × SD)], as determined in Materials and Methods. The ELISA capability evaluations showed that the OD450 values of all 20 negative control sera were under 0.097. Of the 10 positive sera, 3 showed OD450 that ranged from 0.250 to 0.600, 4 were from 0.600 to 1.200, and the remaining 3 were greater than 1.200. In addition, when the sensitivity control sample was diluted 1∶64, it gave an absorbance of 0.132. The CV values of 3 sequential batches of products were 3.69%, 4.11% and 3.96% respectively (n
= 10 for each batch).
In clinical trials, 442 sera of SARS patients and 302 sera of convalescent SARS patients supplied from four hospitals were tested using GST-N2 ELISA, with average positive rates of 70.4% and 75.8%. While 2726 sera from non-SARS patients and healthy individuals were tested and only 2 sera yielded OD values above the cutoff. The false positive rate was 0.07%. After the event, one of the two cases was diagnosed as having SARS. The results for all the clinical samples tested are summarized in Table 4. These results indicated that GST-N2 ELISA provided promising sensitivity and specificity for SARS-CoV antibodies testing.
| Patient type (days after illness) | No. of samples | No. of positive samples | Positive rate (%) |
|---|---|---|---|
| Beijing XiaoTangShan Hospital | |||
| SARS inpatients (0–20) | 14 | 6 | 42.9 |
| SARS inpatients (21–30) | 63 | 41 | 65.1 |
| SARS inpatients (31–40) | 86 | 60 | 69.8 |
| SARS inpatients (41–50) | 93 | 67 | 72.0 |
| SARS inpatients (>50) | 51 | 43 | 84.3 |
| Total | 307 | 217 | 70.7 |
| Health-care workers | 544 | 0 | 0 |
| The PLA General Hospital | |||
| Convalescent SARS patients | 302 | 229 | 75.8 |
| Non-SARS patients | 1558 | 2 | 0.13 |
| Healthy individuals | 616 | 0 | 0 |
| Non-SARS total | 2174 | 2 | 0.09 |
| Beijing Sino-Japen Friendship Hospital | |||
| SARS inpatients (0–20) | 2 | 1 | 50 |
| SARS inpatients (21–30) | 15 | 7 | 46.7 |
| SARS inpatients (31–40) | 23 | 14 | 60.9 |
| SARS inpatients (41–50) | 26 | 19 | 73.1 |
| SARS inpatients (>50) | 34 | 28 | 82.4 |
| Total | 100 | 69 | 69.0 |
| Beijing Hospital of Integrated Traditional Chinese and Western Medicine | |||
| Diagnosed SARS patients | 35 | 25 | 71.4 |
| Probable SARS patients | 8 | 1 | 12.5 |
| Exclusion in final diagnosis | 8 | 0 | 0 |
Discussion
In this report, 16 recombinant SARS-CoV proteins and certain protein fragments, including full-length N protein, nine fragments of N proteins fused to GST, four partials of S proteins and two partials of M proteins fused to TRX, were evaluated for their potential as diagnostic antigens to detect anti-SARS antibodies. It is very dangerous to extract the antigens from inactive SARS-CoV, therefore we used an E. coli expression system to produce these target proteins. Numerous experiments were undertaken to confirm that GST-N2 fusion protein had stable, soluble and abundant expression in E. coli. In contrast to other SARS-CoV structural proteins that contain multiple sites for glycosylation during infection, which may cause different immunoresponses, the N protein is free of glycosylation sites and does not change its immunological characteristics even when expressed in a prokaryotic system.17The serological reactivity of these recombinant proteins was tested by protein microarrays, which were used to screen the most specific antigens for developing the ELISA. The tests showed that although recombinant S protein fragments presented strongly positive results for the antibodies in SARS sera using protein microarray analysis, their non-specific reactivity with the sera from healthy individuals was strong (about 70%). However, GST-N2 fusion protein was weakly non-specific with negative sera, and importantly, 105 out of 122 samples (86.1%) from SARS patients showed positive reactivity against the GST-N2 fusion antigen in clinical assessment. Whereas GST and TRX might lead to non-specific reaction in immunoassay, both positive sera and negative sera were tested by microarray assay. The data revealed that TRX could cross-react with the antibodies in both SARS sera and negative sera, but GST could not. Subsequently, the N protein fragment screening microarray contained an internal dose–response IgG calibration curve that was used to assess the antigenicity of various N fragments. All results from the microarray analysis showed recombinant GST-N2 antigen was the potential candidate for establishment of an ELISA assay. Our data indicate that a protein microarray assay with indirect fluorescence detection is an effective method that can be used to screen the specific antigens through determining the different reactivities of antibodies from serum samples directed against printed antigens. The presence of calibration curves allowed us to demonstrate the amounts of specific antibodies from different serum samples. In this format, any given signals within the arrayed antigens were interpolated to the calibration curve processed under the same conditions as the antibody reactivity being determined. The calibration curves provided the advantage unique of minimizing the matrix effects.
We used GST-N2 fusion protein as a coated antigen to develop an indirect ELISA method and optimized a series of experimental conditions. In clinical trials, positive rates were 70.4% and 75.8% respectively for SARS inpatients and convalescent patients, while only 2 out of 2726 (0.07%) negative sera were tested as false positive. One of these two cases was diagnosed as SARS later. The sensitivity of the GST-N2 ELISA was 1∶64. Reported data showed a high degree of sensitivity and specificity for SARS-CoV antibody testing, which were similar to those reported by Shi et al.18 But then, it is expected that the positive predictive value of GST-N2 ELISA could be improved. Compared with ELISA, microarray assay has a higher positive rate and sensitivity (86.1% and 1∶200). This is reasonable considering the different reaction formats in the two assays. The detection of fluorescence signals emitted from the laser-excited fluorophores on the array is amplified by a photomultiplier tube.
We inferred that there were two reasons causing false negatives in serological testing. Above all, early in the SARS outbreak, it was likely that some non-SARS pneumonic patients mingled with SARS patients as the two could not be distinguished by clinical diagnosis criteria.19 Besides, serology is not particularly helpful in the early-acute-phase of patients as it takes at least 3 weeks from the onset of clinical symptoms to become positive. The data was a mean, which ignored the stages of disease. There has been a lack of a “gold standard” of laboratory methods for SARS diagnosis up to now. SARS-CoV infection wasn't excluded even if a negative signal was presented in antibody tests, according to the notice declared by the Department of General Administration of Ministry of Health, People's Republic of China. It is indicated that antibody testing could be a complementary diagnosis means for SARS disease in clinics and used in epidemiologic investigations, yet it was not suitable to be a preliminary screen of SARS patients.
In addition, our report provided a pilot study about identification of the specific antigenic region of N protein, which would be useful for finding immunoreactive epitopes of N protein. Previous research about SARS-CoV proteins revealed that, in all coronavirus structural proteins, the N protein is highly conserved, immunogenic, and abundantly expressed during infection,20–24 both in mRNA and protein levels.25 Our data from antigen screening microarrays indicated that recombinant N2 protein, which is located at 213aa–423aa of the C-terminus of N protein, exhibited a stronger antigenic reaction than other fragments of N protein. The C-terminus of N protein has a high composition of basic amino acid residues, most of which display hydrophilicity and immunogenicity. Interestingly, deletion of the 11 amino acids from the C-terminus of N protein disrupted the epitope configuration recognized by all of the conformation-dependent monoclonal antibodies.26 This suggested that important immunoreactive epitopes would locate at the C-terminal region of N protein. Studying the immunogenic properties of the C-terminus of SARS-CoV N protein may form the basis for the mechanism of N protein immunological response, and also for vaccine development against SARS disease.
In conclusion, recombinant GST-N2 fusion protein has potential value as a specific antigen for developing clinical SARS antibody testing. Considering its safer, more sensitive and cost-effective characteristics, the ELISA assay could be used in the diagnosis of SARS infection and in epidemiologic investigations. Besides, protein microarrays can provide a rapid, parallel and high-throughput antigen screening platform and there will be room for improvement in the near future.
Acknowledgements
The authors thank Beijing GOLDENWEIKAI Biotechnology CO. LTD. for their kind help with ELISA assays, Dr. Sheng-Li Bi, from China CDC, for giving us some of the spike proteins and Ping Qiao for protein microarray printing. Yu Ding and Yu-Wei Fan are thanked for synthesis of oligonucleotide primers and sequence determination. We are also grateful to Beijing XiaoTangShan Hospital, The PLA General Hospital, Beijing Sino-Japen Friendship Hospital and Beijing Hospital of Integrated Traditional Chinese and Western Medicine for supplying the serum samples for the clinical assessment. The work was supported by National 863 High-Tech project from the Ministry of Science and Technology of the People's Republic of China.References
- S. M. Poutanen, D. E. Low, B. Henry, S. Finkelstein, D. Rose and K. Green, et al., Identification of severe acute respiratory syndrome in Canada, N. Engl. J. Med., 2003, 348, 1995–2005 CrossRef.
- N. Lee, D. Hui, A. Wu, P. Chan, P. Cameron and G. M. Joynt, et al., A major outbreak of severe acute respiratory syndrome in Hong Kong, N. Engl. J. Med., 2003, 348, 1986–1994 CrossRef.
- J. S. M. Peiris, S. T. Lai, L. L. M. Poon, Y. Guan, L. Y. C. Yam and W. Lim, et al., Coronavirus as a possible cause of severe acute respiratory syndrome, Lancet, 2003, 361, 1319–1325 CrossRef CAS.
- T. G. Ksiazek, D. Erdman, C. Goldsmith, S. R. Zaki, T. Peret and S. Emery, et al., A novel coronavirus associated with severe acute respiratory syndrome, N. Engl. J. Med., 2003, 348, 1953–1966 CrossRef CAS.
- P. A. Rota, M. S. Oberste, S. S. Monroe, W. A. Nix, R. Campagnoli and J. P. Icenogle, et al., Characterization of a novel coronavirus associated with severe acute respiratory syndrome, Science, 2003, 300, 1394–1399 CrossRef CAS.
- M. A. Marra, S. J. M. Jones, C. R. Astell, R. A. Holt, A. Brooke-Wilson and Y. S. N. Butterfield, et al., The genome sequence of the SARS-associated coronavirus, Science, 2003, 300, 1399–1404 CrossRef CAS.
- E. D. Qin, Q. Y. Zhu, M. Yu, B. C. Fan, G. H. Chang and B. Y. Si, et al., A complete sequence and comparative analysis of a SARS-associated virus (Isolate BJ01), Chin. Sci. Bull., 2003, 48, 941–948 Search PubMed.
- L. Enjuanes, C. Smerdou, J. Castilla, I. M. Anton, J. M. Torres and I. Sola, et al., Development of protection against coronavirus induced disease, Adv. Exp. Med. Biol., 1995, 380, 197–211 CAS.
- C. L. Yeager, R. A. Ashmun, R. K. Williams, C. B. Cardellichio, L. H. Shapiro and A. T. Look, et al., Human aminopeptidase N is a receptor for human coronavirus 229E, Nature, 1992, 357, 420–422 CrossRef CAS.
- C. A. M. de Haan, M. Smeets, F. Vernooij, H. Vennema and P. J. M. Rottier, Mapping of the coronavirus membrane protein domains involved in interaction with the spike protein, J. Virol., 1999, 73, 7441–7452 CAS.
- A. Lueking, M. Horn, H. Eickhoff, K. Bussow, H. Lehrach and G. Walter, Protein microarrays for gene expression and antibody screening, Anal. Biochem., 1999, 270, 103–111 CrossRef CAS.
- G. MacBeath and S. L. Schreiber, Printing proteins as microarrays for high-throughput function determination, Science, 2000, 289, 1760–1763 CAS.
- L. Mezzasoma, T. Bacarese-Hamilton, M. Di Cristina, R. Rossi, F. Bistoni and A. Crisanti, Antigen microarrays for serodiagnosis of infectious diseases, Clin. Chem., 2002, 48, 121–130 CAS.
- J. Sambrook and D. W. Russell, Molecular Cloning: A laboratory manual, Cold Spring Harbor Laboratory Press, New York, 3rd edn., 2001 Search PubMed.
- M. Schena, D. Shalon, R. W. David and P. O. Brown, Quantitative monitoring of gene expression patterns with a complementary DNA microarray, Science, 1995, 270, 467–470 CrossRef CAS.
- D. Shalon, S. J. Smith and P. O. Brown, A DNA microarray system for analyzing complex DNA samples using two-color fluorescent probe hybridization, Genome Res., 1996, 6, 639–645 CrossRef CAS.
- M. C. Mechin, M. Der Vartanian and C. Martin, The major subunit ClpG of Escherichia coli CS31A fibrillae as an expression vector for different combinations of two TGEV coronavirus epitopes, Gene, 1996, 179, 211–218 CrossRef CAS.
- Y. L. Shi, Y. P. Yi, P. Li, T. J. Kuang, L. H. Li and M. Dong, et al., Diagnosis of severe acute respiratory syndrome (SARS) by detection of SARS coronavirus nucleocapsid antibodies in an antigen-capturing enzyme-linked immunosorbent assay, J. Clin. Microbiol., 2003, 41, 5781–2 CrossRef CAS.
- K. L. Hon, A. M. Li, F. W. Cheng, T. F. Leung and P. C. Ng, Personal view of SARS: confusing definition, confusing diagnosis, Lancet, 2003, 361, 1984–1985 CrossRef CAS.
- L. Kuo and P. S. Masters, Genetic evidence for a structural interaction between the carboxy termini of the membrane and nucleocapsid proteins of mouse hepatitis virus, J. Virol., 2002, 76, 4987–4999 CrossRef CAS.
- K. Narayanan, C. J. Chen, J. Maeda and S. Makino, Nucleocapsid-independent specific viral RNA packaging via viral envelope protein and viral RNA signal, J. Virol., 2003, 77, 2922–2927 CrossRef.
- L. Yu, W. Liu, W. M. Schnitzlein, D. N. Tripathy and J. Kwang, Study of protection by recombinant fowl poxvirus expressing C-terminal nucleocapsid proteins of infectious bronchitis virus against challenge, Avian Dis., 2001, 45, 340–348 Search PubMed.
- J. A. Hiscox, T. Wurm, L. Wilson, P. Britton, D. Cavanagh and G. Brooks, The coronavirus infectious bronchitis virus nucleoprotein localizes to the nucleolus, J. Virol., 2001, 75, 506–512 CrossRef CAS.
- C. Liu, T. Kokuho, T. Kubota, S. Watanabe, S. Inumaru and Y. Yokomizo, et al., DNA mediated immunization with encoding the nucleoprotein gene of porcine transmissible gastroenteritis virus, Virus Res., 2001, 80, 75–82 CrossRef CAS.
- M. M. Lai and D. Cavanagh, The molecular biology of coronaviruses, Adv. Virus Res., 1997, 48, 1–100 Search PubMed.
- S. K. Wootton, E. A. Nelson and D. Yoo, Antigenic structure of the nucleocapsid protein of porcine reproductive and respiratory syndrome virus, Clin. Diagn Lab. Immunol., 1998, 5, 773–779 CAS.
| This journal is © The Royal Society of Chemistry 2005 |