Optical calcium sensors: development of a generic method for their introduction to the cell using conjugated cell penetrating peptides
Abigail Webstera, Steven J. Comptonb and Jonathan W. Aylott*c
aDepartment of Chemistry, University of Hull, Hull, UK HU6 7RX. E-mail: a.webster@chem.hull.ac.uk; Fax: +44 (0)1482 466410; Tel: +44 (0)1482 465484
bRespiratory Medicine, The Vision of Academic Medicine, Castle Hill Hospital, Cottingham, Yorkshire, UK HU16 5JQ. E-mail: s.j.compton@hull.ac.uk; Tel: +44 (0)1482 466034
cSchool of Pharmacy, The University of Nottingham, Nottingham, UK NG7 2RD. E-mail: Jon.Aylott@nottingham.ac.uk; Fax: +44 (0)115 9515102; Tel: +44 (0)115 9516229
First published on 15th December 2004
Abstract
Probes Encapsulated By Biologically Localised Embedding (PEBBLEs) are optical sensors with nanometer dimensions fabricated by microemulsion polymerisation. The most beneficial characteristic of these sensors is the protection offered by the sensor matrix which decreases interaction between the fluorophore and the cell. These sensors have been introduced to the cell by a number of methods; however this paper discusses the development of a generic method to facilitate inclusion of this type of sensor in the cell by a simple incubation step. This was achieved by covalent linkage of a synthetic Cell Penetrating Peptide (CPP) based on the Human Immuno-deficiency Virus (HIV) -1 Tat, to the external sensor matrix. Calcium sensors were used to demonstrate this approach to incorporate the sensors within the cell. Characterisation revealed the calcium sensors were approximately 30 ± 7 nm in diameter with a slightly negative zeta potential. The sensors demonstrated a linear range of 0–50 µM with negligible interference from a range of cellular ions and protein. Leaching of entrapped dyes from the calcium sensors was determined as 3% in a 24 h period, while photobleaching of the entrapped dye was minimal over a 40 min period. The sensors ability to cross the cell membrane using the covalently attached synthetic Tat peptide is demonstrated. Cellular inclusion of the sensors occurred within a 30 min incubation period.
Introduction
Cell penetrating peptides are described as peptides with a maximum of 30 residues able to cross the cell membrane in a seemingly energy independent manner.1 Frequently investigated CPPs include buforin from Bufo bufo gargarizans, penetratin from Drosophilia, arginine rich peptides and Tat from the virus HIV. A common feature of all these peptides is their cationic nature which has been shown to attach strongly to the cell membrane.2Frankel first suggested Tat as a vehicle for protein delivery in 1991,3 though it was not until 1997 that this hypothesis was successfully realised, when a suitable technique was developed by Dowdy and coworkers.4 Overcoming the barrier of the cell membrane to deliver membrane impermeable cargos often requires harsh methods such as electroporation or liposomal transfection which display a number of shortcomings including unwanted cellular effects and their limitation to in vitro applications.1 However, CPPs have been used to deliver a range of cargos including proteins,5 drugs6 and nanoparticles7,8 into the cell with no discernible cellular effects or toxicity issues.1,9
The mechanism for the uptake of CPPs has been extensively debated1,10 and recent investigations have shown significant endocytotic contribution to the cellular uptake of CPPs,11 with ionic interactions at the cell surface preceding endocytotic uptake.12 Although there are a number of scientifically viable proposals for cellular entry, there is still no definitive mechanism for CPPs and the mode of cell entry may be highly dependant on the peptide, its cargo and the cell type.10 As a delivery vector they offer a number of appealing properties including cellular targeting and effective in vitro delivery of cargos with no apparent restriction of size, hydrophobicity restraints or cell type and without antigenic, immunogenic, inflammatory properties or disturbance to the cell membrane.1 Eventual location of the cargo is dependant on the CPP, its cargo and the type of attachment used to bind the two. Targeting of particular organelles can be accomplished by the addition of further address motifs.10
Calcium is one of the most important second messengers in the body with roles in both intracellular and intercellular communication.13 It is a highly versatile signalling ion able to regulate the coordination of a range of vital and diverse functions. Free calcium can be highly heterogeneous within different cells, with concentrations ranging from nanomolar to micromolar in the resting cytoplasm and endoplasmic reticulum respectively.14 Calcium stores in the endoplasmic reticulum allow mobilisation of cellular calcium under the control of inositol-1,4,5-trisphosphate,15 permitting propagation of calcium signals to move from cell to cell in a wave-like motion.13 Calcium is able to achieve this fine balance between signalling ‘on and off’ reactions via the combined action of buffers, pumps and exchangers.16
Many probes exist to quantify calcium in the cellular environment with fluorescent dyes providing the most established method.17 There are numerous calcium sensitive dyes available and considerable literature exists on their characterisation and fluorescence properties both in vivo and in vitro, which has been reviewed.18 However, fluorescent dyes used for monitoring calcium in cells experience a number of inherent difficulties that can lead to erroneous results such as the inaccurate estimation of calcium concentrations.14 Complications encountered using fluorescent dyes include interference from other cellular ions or non-specific protein binding of the dye, which result in fluorescence intensity changes without a concomitant variation of calcium concentration. Entry of fluorescent probes into the cellular environment is often facilitated by the acetoxymethyl (AM) ester attached to the fluorophore. Production of an active fluorophore requires cleavage of the AM ester by esterase enzymes within the cell. Cytotoxicity is a consequence of the production of an active fluorophore because one of the by-products of this cleavage is formaldehyde,14 while incomplete hydrolysis of the fluorophore results in molecules of inactive dye. Leakage of the dye from the cell is a result of the dye being extruded by P-glycoprotein multidrug transporters or by ionic transporters; this effect can be prevented by chemicals such as sulfinpyrazone, although this inhibits the cellular response.19,20 These difficulties as well as the cytotoxic issues associated with the use of fluorescent dyes indicate that long-term monitoring of calcium within a cell will be difficult to achieve. Calibrations produced in vitro are not necessarily valid in vivo because of these issues.14
Entrapment of fluorescent dyes in PEBBLEs offers a number of benefits that cannot be obtained when using the free dyes. Firstly the sensor matrix protects the fluorescent dye from cellular interferences, for example the non-specific binding of the dye by cellular proteins as the pores of the matrix prevents cellular interaction with the fluorophore. Sequestering of the dye by organelles is prevented by the physical size of the sensors. The sensor matrix reduces the cytotoxic effect of the fluorophore primarily because the AM ester is not required to deliver the dye into the cell but fundamentally because the dye is not in contact with the cellular contents.21 Another advantage of using PEBBLEs is that ratiometric sensors can be produced using dyes which are not normally ratiometric, in this case the co-immobilisation of the calcium sensitive dye fluo-4 with the reference dye alexa fluor 568. Ratiometric sensors provide a means to negate erroneous alterations in the fluorescence signal, caused by environmental factors such as variation in temperature, focal change shifts or photobleaching. In addition, the fabrication process confers homogeneous distribution of the entrapped dyes within the sensors allowing spatial resolution and continuity to ratiometric measurements in vitro.22
A crucial factor when fabricating sensors for intracellular measurements is the size of the sensor, as physical disruption of the cell can induce spontaneous cell lysis. PEBBLEs range in diameter from 20–300 nm which is small enough to cause no perturbations to either the cell or cell membrane that could stimulate apoptosis.23 The method used for the production of these sensors is very versatile using biologically inert matrices such as sol gel,24 polyacrylamide25 or liquid polymers26 allowing both entrapment within and functionalisation of the sensor. PEBBLEs have been produced for a range of cellular analytes including glucose27 and calcium.21 Entrapment of analyte recognition components within the matrix enables complex sensor production; for example the glucose sensor in which glucose oxidase is located in close proximity to an oxygen sensitive fluorophore,27 thus exploiting the fact that the reaction between glucose and oxygen is 1 ∶ 1, to measure glucose by fluorescence utilising the quantifiable oxygen sensitive characteristics of a ruthenium fluorophore.
One of the main issues encountered using PEBBLEs is their introduction to the intracellular environment without irreversibly damaging the cell membrane. A number of methods have been employed by Kopelman and co-workers to deliver PEBBLEs into the cell; these include gene gun, liposomal transfection, micropipette and phagocytosis.25 The aim of this work is to extend the range of options for transporting these sensors to the cell by developing a generic delivery method that uses a simple incubation step. In order to achieve this, we functionalised the outer surface of calcium specific polyacrylamide sensors with a synthetic CPP (CaCPP). The CPP chosen for this work is an 11 residue synthetic peptide based on HIV Tat. Tat is an 86 residue transcriptional regulator which is essential for the replication of HIV; CPP functionality has been located in residues 47–60 of the basic domain,28 although peptide lengths of fewer residues have been found to retain this characteristic. The ability of the CaCPP sensors to translocate mammalian cell membranes will be demonstrated.
Methods and materials
Reagents
Unless stated all reagents, solvents and buffers were purchased from Aldrich (Dorset, UK). Water with a purity of 5 M Ω cm was prepared by reverse osmosis and ion exchange using an Elgastat Option 4 water purifier (Elga Ltd, High Wycombe, Bucks, UK).Dyes
The fluorescent indicators Fluo-4 and Alexa Fluor 568 attached to 10,000 MW dextran purchased from Molecular Probes (Leiden, The Netherlands) were diluted to 5 mg ml−1 and stored in frozen aliquots.Production of polyacrylamide calcium sensors
Calcium sensors were produced using a modified method of Clarke et al.21 The acrylamide solution consisted of 18.2% acrylamide, 5.9% N,N-methylenebis (acrylamide) 1.9% N-(3-aminopropyl) methacrylamide hydrochloride (Polysciences Inc, Warrington, UK) in 1.8 ml of 10 mM 3-(N-Morpholino) propanesulfonic acid (MOPS) buffer (Sigma, Gillingham, Dorset, UK)/100 mM KCl (BDH Laboratory Supplies, Poole, UK) at pH 7.4. 1.8 ml of acrylamide solution was used with a maximum of 40 µl of the fluorescent reporter Fluo-4 and 10 µl of the reference dye Alexa Fluor 568. A final aqueous volume of 2 ml was achieved by the addition of 10 mM MOPS/100 mM KCl buffer.Water in oil microemulsion polymerisation was used to produce the sensors; the microemulsion consisted of an oil phase of deoxygenated hexane (42 ml) and a surfactant phase of sodium dioctylsulfosuccinate (0.08 M) and Brij 30 (0.19 M), to which a total aqueous phase of 2 ml was added. Polymerisation was initiated with sodium metabisulfite (SMBS) (10% w/v 10 µl) and the reaction was allowed to proceed for 2 h with stirring in an argon environment. At completion of polymerisation, hexane was removed by rotary evaporation and the sensors were precipitated in absolute ethanol. Surfactants were removed with absolute ethanol and the sensors were collected by centrifugation. Excess ethanol was removed by filtration (Whatman Anodisc 25, 0.02 µm filter) and the sensors were dried under vacuum in a desiccator at 4 °C.
Functionalisation of sensors
Sensors were functionalised with a custom 11 residue peptide sequence CRRRQRRKKRG (Sigma-Genosys, Cambridge) with a final purity of 95%. Covalent attachment of the peptide to the sensor was accomplished using a modified method originally described by Shriver-Lake29via the heterobifunctional crosslinker γ-maleimidobutyric acid N-hydrosuccinimide ester (GMBS). Scheme 1 summarises the reaction chemistry for the attachment of the peptide to the sensor; the method will be described in brief.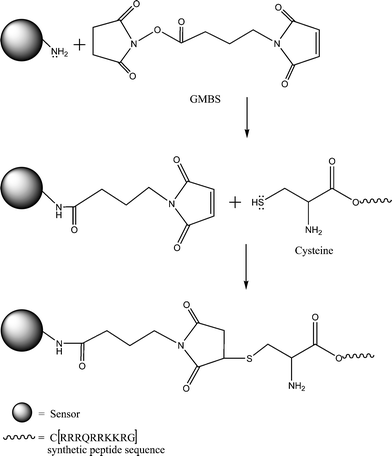 | ||
| Scheme 1 Schematic of immobilization chemistry illustrating GMBS linking of the cysteine residue on the synthetic Tat peptide to the outer matrix of a polyacrylamide sensor. The peptide sequence represented by the symbols of the amino acids used where C = cysteine, R = arginine, Q = glutamine and K = lysine. | ||
An aliquot of calcium sensors was immersed in 2 mM GMBS in ethanol with agitation for 1 h. Excess GMBS was removed by washing in 20% H2O in ethanol; functionalised sensors were collected by centrifugation. 5 mg synthetic Tat peptide was dissolved in 5 ml of 10 mM MOPS/100 mM KCl buffer. This solution was added to the GMBS functionalised sensors and agitated for 2 h at room temperature, then overnight at 4 °C. Unbound peptide was removed by washing in 20% H2O in ethanol and the CaCPP sensors were collected by centrifuge, filtered with ethanol and dried under vacuum in a desiccator at 4 °C.
Fluorescence data
Measurements for calibration and characterisation studies were collected on AB2 version 4 software using an Aminco Bowman Series 2 Luminescent Spectrometer with continuous wave 150 W xenon lamp. Samples were prepared using 5 or 10 mg of sensors in 2 ml of 10 mM MOPS/100 mM KCl buffer in UV grade disposable cuvettes. Calcium standards in 10 mM MOPS/100 mM KCl buffer were calibrated using a calcium ion selective electrode (Spectronic Analytical Instruments, Garforth, Leeds, UK).Fluorescence imaging
The transformed human epithelial cell line NCTC 2544 was used for fluorescence imaging. Cells were incubated with 0.5 mg CaCPP sensors in 100 µl PBS for 0.5–4 h, 24 h after cells had been passaged onto 3 cm Petri dishes. Sensors which had not been internalised by the cell were removed with PBS washes prior to removal of the cover disk for fluorescence imaging. Fluorescence imaging was performed on a Leica DM IR microscope using a 40X objective lens; excitation was achieved using a Hg 50 W lamp with a CHROMA FITC filter cube, images were produced with a Diagnostic Instruments SPOT slider 2 camera.Lysing studies
Wild type Chinese hamster ovary (CHO) cells expressing hPAR2 were incubated for 4 h with aliquots of CaCPP sensors and calcium sensors. Non-internalised sensors were removed by washing with PBS, prior to detaching cells using 5 mM EGTA in PBS; cells were centrifuged and resuspended in fresh media. 100 µl aliquots of cells were placed in cuvettes containing 3 ml PBS doped with 1 mM calcium. The cuvette was placed in the fluorimeter and the baseline monitored for several seconds before 100 µl 5 mM Tween 20 was added to the cuvette. Fluorescence monitoring of the lysing studies was performed on a Photon Technology International (PTI) fluorimeter (New Jersey USA), lamp power source −220B. The cells in the cuvette were kept in suspension during fluorescence monitoring by an internal stirrer MD-5020 PTI and data was collected using FELIX PTI V1.426 software.Transmission electron microscope (TEM) studies
CHO cells expressing hPAR2 were fixed in 1% glutaraldehyde in 0.1 M sodium cacodylate buffer pH 7.4 within 2 h of the cells being removed from the incubation flask. The cells remained in fixative for several days prior to embedding. Primary fixative was removed by immersing samples in 1% osmium tetroxide in 0.1 M sodium cacodylate buffer prior to embedding. Samples were embedded in stock resin and sectioned on a Huxley MK2 ultra-microtome using a glass knife. Sections were placed on 75 mesh formvar grids and stained with uranyl acetate for 15 min and lead citrate for 2 min, prior to visualisation on a JOEL 100C TEM (Tokyo, Japan).Characterisation of calcium sensors
Sizing and surface potential
A Malvern Zetasizer 3000HS (Malvern Instruments, Worcs., UK) was used for both sizing of the calcium sensors, achieved by photon correlation spectroscopy (PCS) and zeta characteristics; both experiments were performed in 1 mM NaCl.Calibration and interference studies
Calibration of 5 mg ml−1 sensors in 10 mM MOPS/100 mM KCl buffer was performed in UV grade disposable cuvettes. Ionic interference studies were performed for sodium and magnesium. These studies were carried out using 5 mM of both NaCl and MgCl2 (AnalaR grade reagents BDH Laboratory Supplies, Poole, UK) in 10 mM MOPS/100 mM KCl buffer with 2.5 mg ml−1 sensor or an equivalent amount of free dye in a cuvette. After addition of ionic interferents, aliquots of calcium standard were added by serial addition. Protein interference was performed using a total of 5 mM bovine serum albumin (BSA). Aliquots of calcium standard were added to the cuvette as described for ionic interference testing and changes in the fluorescence intensity were monitored after each addition.Leach data
Leaching experiments were performed using size 5 Tnf DA 24/32 MWCO 12-14000 Da (Medicell International, London, UK) dialysis tubing washed to manufacturers recommendations with 10 mM EGTA in 10 mM MOPS/100 mM KCl buffer. 20 mg of calcium sensors were placed inside a dialysis tubing lined plastic bottle with a 10 ml aliquot of 10 mM MOPS/100 mM KCl buffer (5 ml either side of dialysis tubing) and agitated for 24 h on a flask shaker (SFI Stuart Scientific, UK) at 500 oscillations min−1. Fluorescence measurements of the solutions from the inner and outer dialysis tubing were taken and calculations were performed to give % values for leached fluorescent dyes.Photobleaching
Photobleaching experiments were performed on both free fluo-4 and calcium sensors using 5 mg ml−1 sensors or an equivalent amount of free dye in UV grade disposable cuvettes in 2 ml of 10 mM MOPS/100 mM KCl buffer. The samples were monitored using continuous excitation at 488 nm and fluorescence data was collected at 1 s intervals for a period of 40 min.Results and discussion
Sensor characterisation
Characterisation and calibration are important elements of producing sensors although calibrations performed in vitro are not necessarily valid in vivo or even from one cell line to another. The calcium sensors were observed to be optically clear when placed in suspension at concentrations as high as 30 mg ml−1 in 10 mM MOPS/100 mM KCl buffer. Calcium sensors were investigated for linearity and dynamic range both with and without the presence of cellular interferents (ionic and protein) allowing for the observation of the sensors response to expected cellular interferents. These calibrations were also compared with the linear range and the response of free dye to the same interferents.The sensors were characterised for size and zeta potential because it is known that the combined effects of zeta potential and size can give insight into the stability of particles in solution. This stability is an important characteristic for the calibration of the sensors in assay as the sensors need to remain homogeneously distributed throughout the cuvette during fluorescence monitoring to ensure continuity of response during serial additions of calcium.
Leaching of the fluorophores from the sensors was studied because high leaching would negate the benefits gained from entrapping the dyes. Leaching of the dye would have two negative effects (1) the cytotoxicity of the free dyes in the cellular environment could interfere with normal cellular functioning, which over prolonged periods can induce apoptosis and (2) the fluorescence signal of the dyes are changed by the presence of protein which would have a direct effect on the fluorescence response.
Linearity and dynamic range
Linearity and dynamic range were determined using a ratio of the reference dye and the calcium fluorophore, which enabled the production of a calibration graph that was not subject to fluctuations caused by the assay environment such as dilution effects caused by the addition of calcium standard. The calcium sensors demonstrated excellent sensitivity to increasing concentrations of calcium, with large changes in fluorescence intensity confirmed by the slope of the graph which can be seen in Fig. 1. The linear range of the calcium sensors was determined as 0–50 µM, which is a suitable range for cellular measurements, while the lower limit of detection was determined as 0.085 µM, calculated from the variance around the line. | ||
| Fig. 1 Calibration graph showing the linearity of 10 mg of calcium sensors in 2 ml MOPS/KCl buffer, to serial addition of calcium. The graph is a ratio of the fluorescence intensity of the calcium chelating dye fluor-4 (R1) divided by the reference by alexa fluor 568 (R2). The linear range was found to be 0–50 µM and the lower limit of detection was 0.085 µM. | ||
Size and zeta potential
The size of the sensors was found to be approximately 30 ± 7 nm in diameter determined by PCS. The CaCPP sensors retained their optical clarity at concentrations identical to the non-functionalised sensors in PBS. The sensors demonstrated a slightly negative zeta potential of approximately −25 mV in 0.1 mM NaCl, which is not considered sufficient to maintain the sensors in suspension for extended periods. However, it was observed that the sensors were able to retain suspension for prolonged periods of greater than 7 days. The literature states that the stability of aqueous particle dispersions due to electrostatic interactions require zeta potential values of greater than ±30 mV30 therefore the continued suspension of the particles was most likely an effect of the size and concentration of the sensors in solution rather than electrostatic interactions of the outer sensor matrix.Interference study
The interference study was used to determine how the fluorescence response of the fluo-4 entrapped within the sensors may be altered by the presence of ionic and protein interference; this data was compared to the response of free fluo-4 to similar concentrations of the same interferents. Aliquots of BSA or ionic interferent were added to solutions of either PEBBLEs or free dye. The changes in fluorescence intensity were monitored for the additions of the interferents. The sensors in solution displayed no change in fluorescence intensity to the addition of BSA whereas the solution of free dye showed a steady increase in fluorescence intensity with each addition of BSA. This difference in observed response to BSA between free dye and sensors was a result of the matrix which encapsulates the dye of the sensors. The pores of the matrix are too small to allow the BSA access to the internal portion of the sensor and consequently protect the entrapped dyes from protein interference. Both sensors and free dye reacted similarly with the addition of ionic interferents, displaying a small decrease in fluorescence signal during the addition of ionic interferent. The similarity of these results is again due to the pores of the matrix which will allow free access of ions to the internal sensor matrix.After the addition of interferents the fluorescence response of both the free dye and sensors to increasing calcium concentration was plotted in Fig. 2a and 2b. The free dye containing BSA demonstrated an increased fluorescence intensity to the initial addition of calcium after which almost no change in fluorescence intensity was observed to increasing calcium concentrations. In comparison, the sensors displayed an increasing fluorescence signal to increasing calcium concentrations. There was a slight reduction in linearity when calcium concentrations were above 40 µM with some loss of sensitivity when compared with the calibration graph (Fig. 1). However, these changes were not as great or as severe as the changes seen in the free dye to the same levels of interferents; this response can be seen in Fig. 2a. The response of both free dye and sensors to ionic interference demonstrated the sensors retained linearity with a slight loss of range when the calcium concentration was above 40 µM, whereas the free dye exhibited a reduction in sensitivity and linearity when compared with the sensors; this data can be seen in Fig. 2b.
 | ||
| Fig. 2 (a) Plot of interference testing for 5 mg sensors or equivalent free dye in 2 ml MOPS/KCl buffer. Protein testing was performed using 5 mM bovine serum albumen prior to the serial addition of calcium. The graph illustrates the loss of linearity and response for the free dye when compared with the calcium sensors, which were not excessively affected by the presence of the BSA. (b) Plot of interference testing for 5 mg sensors or equivalent free dye in 2 ml MOPS/KCl buffer, both samples were subjected to additions of a solution of Mg and Na prior to the serial addition of calcium. The graph compares the fluorescence response of both the sensors and free dye to serial additions of calcium. The sensors on the whole retain their sensitivity and linearity; however the free dye demonstrates a loss of both linearity and sensitivity to calcium in the presence of ionic interferents. | ||
Functionalisation of the sensors
The calcium sensors were functionalised using the protocol described in the methods section of this paper. CaCPP sensors were functionalised with a custom 11 residue peptide based on the HIV-1 Tat CPP described by Caron et al.;28 however a cysteine residue was added during peptide synthesis to enable covalent linking of the peptide to the outer sensor matrix. The heterobifunctional crosslinker GMBS achieved covalent linkage between the thiol group on the cysteine residue of the synthetic peptide with amine groups on the sensor matrix; linkage chemistry can be seen in Scheme 1.Leaching and photobleaching study
This study determined the total amount of dye leaching from the sensors as 3% in a 24 h period. The sensors and free dye were subjected to continuous excitation at 488 nm for a period of 40 min and the fluorescence response was taken every second. The free dye displayed the effects of photobleaching within 3.5 min whereas the sensors displayed virtually no reduction in fluorescence intensity over the total time period of 40 min. It is not apparent why the sensors exhibit reduced photobleaching when compared to the free dye as it would be expected that, due to the optical quality of the matrix used to entrap the fluorophore, both free and entrapped dye would photobleach similarly. However it is possible that the polyacrylamide sensor matrix imparts enhanced stability to the excited state fluorophore, hence lessening the amount of photobleaching observed in the sensors. Although we cannot fully identify the origins of this effect, Clark et al.21 also reported this characteristic and indeed we have observed this effect with other matrix-entrapped fluorophores.Cell studies
CaCPP sensors were incubated with the transformed human epithelial cell line NCTC 2544 for between 0.5–24 h. The cells demonstrated no perturbation to the shortest incubation time and showed no signs of cytotoxicity when incubation times were extended up to 24 h, with the cells continuing to proliferate normally, which is in agreement with the literature.25,27 The cells displayed increased fluorescence upon the addition of calcium ionophore which causes rapid equilibration of intracellular and extracellular calcium concentrations; the observed fluorescence can be seen in Fig. 3. | ||
| Fig. 3 Fluorescence image of CaCPP sensors in the transformed human epithelial cell line NCTC 2544 after a 30 min incubation period. The image shows the fluorescence in the cytoplasm around the nucleus of the cells, caused by sensors transported through the cell membrane by the covalently attached CPP; the fluorescence signal was produced by the addition of calcium ionophore to the cells prior to imaging. | ||
Cells which had been incubated with calcium sensors that did not possess CPP functionality did not display the increase in fluorescence intensity seen for the CPP calcium sensors. The fluorescence intensity observed with the cells indicated CaCPP sensors were not being removed by the washing procedure and in addition that the sensors remained active during the extended incubation times. However these observations did not establish whether the increase in fluorescence signal was caused by the sensors being attached to the surface of the cells via the peptide or if the sensors had gained access to the cytoplasm.
Experiments were performed on CHO cells to assess the ability of the CaCPP sensors to cross the cell membrane of another mammalian cell line using the CPP covalently attached to the exterior of the sensor matrix. A 2–3 fold increase in the baseline fluorescence was observed in cells incubated with CaCPP sensors when compared to either a cell blank with no incubation of sensors or to cells incubated with calcium sensors which did not have the CPP attached. The cell blank and cells incubated with calcium sensors without the CPP attached displayed a similar fluorescence baseline, implying the washing procedure was adequate to remove calcium sensors not functionalised with the CPP.
Lysing studies were used to determine whether the increased fluorescence signal was caused by the CaCPP sensors gaining access to the cell or whether the fluorescence signal was caused by the sensors becoming attached to the outer cell membrane by ionic interactions at the cell membrane. An aliquot of CHO cells was incubated for 4 h with CaCPP sensors in a buffered calcium environment. The cuvette containing the cells was monitored continuously by fluorescence while Tween 20 was used to rupture the cellular membrane. The calcium in the buffer would cause any sensors attached to the outer membrane to be fluorescing prior to rupturing of the cell membrane. Hence any increase in fluorescence intensity could only be attributed to sensors which had been internalised within the cell. This experiment was repeated with equivalent amounts of cells that had been incubated with non-functionalised sensors.
The result of lysing the cell membrane was an increase in fluorescence intensity for the cells incubated with CaCPP sensors, which was not observed in the cells incubated with calcium sensors that did not possess the CPP functionality. The results shown in Fig. 4 demonstrate the increase in fluorescence intensity caused by the release of CaCPP sensors from the cell as the cellular membrane fragmented in response to the addition of the detergent Tween 20. This finding signified the CaCPP sensors were being transported across the cellular membrane by the covalently attached CPP.
 | ||
| Fig. 4 Lysing study of CaCPP and non-functionalised sensors; the graph demonstrates the change in fluorescence when CHO cells incubated with CaCPP sensors were lysed with Tween 20 in a calcium doped buffer (solid line). When a sample of cells incubated with non-functionalised sensors was lysed with Tween 20, no change in fluorescence intensity was observed (broken line). Addition of 5 mM Tween 20 was at approximately t = 50 s for both samples. Incubation time with sensors was 4 h prior to use of the cells in the lysing experiment. | ||
Neither the CHO or the NCTC 2544 cell lines exhibited signs of distress to the inclusion of the sensors into the cytoplasm which is in accord with the literature on this type of sensor.25,27 The cells also displayed no indication of toxicity caused by the CPP, which is again consistent with the literature on both Tat derived peptides and CPPs generally.1,7
TEM
TEM was used to conclusively substantiate the sensors' ability to utilise the CPP functionality of the synthetic peptide to traverse the cell membrane. The micrograph (Fig. 5) shows sensors that have crossed the cell membrane. Sensors that had been transported through the cell membrane were found only in the cytoplasm and not in the nucleus or any organelle which concurs with the literature which states that CPP linked cargos remain in the cytoplasm unless they have been targeted to subcellular locations or organelles by the addition of extra address motifs.10 As there were no address motifs added to the synthetic peptide used in this experiment it was anticipated that the cargo would remain in the cytoplasm.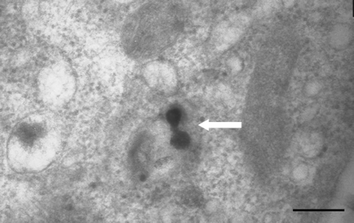 | ||
| Fig. 5 Micrograph depicting two CaCPP sensors in the cytoplasm of a CHO cell, indicated by the white arrow. The sensors were introduced to the cell during a 30 min incubation period, entering the cell using the CPP functionality attached to the outer sensor matrix (size bar = 100 nm). | ||
The size and uniformly spherical shape of the CaCPP sensors was also confirmed by the TEM, though it was not possible to verify whether any of the sensors displayed in the micrographs were the total sensor diameter because the random nature of the sectioning process would mean there were a number of sensors cut at differing portions through the sensor whole. The maximum size of the sensors observed by TEM was approximately 35 nm validating the results of the PCS studies and corroborating the uniformity of the peaks seen in subsequent runs.
Conclusion
The work has successfully demonstrated an innovative procedure for the introduction of PEBBLEs to cells, which utilised the CPP functionality of a synthetic peptide based on 10 residues derived from the basic domain of HIV-1 Tat. Incubation of the sensors in two cell lines over a maximum period of 24 h revealed no discernible impairment to the cells from either the entry of the sensors or cytotoxic effects from the peptide or the entrapped fluorescent dyes.The CaCPP sensors were capable of translocating the cellular membrane within 30 min, however longer incubation times (24 h) with CaCPP sensors caused no observable perturbations to cell functioning with the inclusion of the sensors into the cytoplasm. These findings demonstrate that long-term monitoring of normal calcium fluxes in vitro would be possible using CaCPP sensors. Attachment of the CPP to the sensors has provided a practical technique to introduce these sensors to the cell via a simple incubation step. This method of delivery could have potential applications to all aspects of cell research, especially where the cell line is sensitive to manipulations.
The calcium sensors displayed a linear detection range in the region of 0–50 µM with a lower limit of detection of 0.085 µM. The sensors displayed a slight negative zeta potential and the size of the calcium sensors was determined using PCS to be approximately 30 ± 7 nm in diameter. Interference studies revealed the sensors responded more favourably than the free dye while in the presence of ionic and protein interferents. The sensors retained linearity and dynamic range with increasing calcium concentrations better than the free dye, which showed a marked reduction in both sensitivity to calcium and dynamic range. These findings indicate that calibration graphs produced in vitro may be valid in vivo, which will provide a significant step toward measuring true biological calcium levels. Leaching of dye from the sensors was determined as 3% in 24 h, while photobleaching was negligible for a 40 min period of continuous excitation. Future work will include quantitatively monitoring calcium fluxes for extended periods in a range of cell lines using the CaCPP sensors.
Acknowledgements
The authors would like to thank A. Botham for the CHO hPAR2 cell line, J. Halder for help with the TEM work, H. Savoie for help with the fluorescence imaging and use of the NCTC 2544 cell line. Dr A. D. Walmsley and Dr R. A. Wheatley for discussions and proof reading, and the EPSRC for the DTA, grant number GR/P00031/01 for funding this project.References
- P. Lundberg and U. Langel, J. Mol. Recognit., 2003, 227–233 CrossRef CAS.
- J. A. Leifert and J. L. Whitton, Mol. Ther., 2003, 8, 13–20 CrossRef CAS.
- B. J. Calnan, S. Biancalana, D. Hudson and A. D. Frankel, Genes Dev., 1991, 5, 201–210 CrossRef CAS.
- S. A. Ezhevsky, H. Nagahara, A. M. VoceroAkbani, D. R. Gius, M. C. Wei and S. F. Dowdy, Proc. Natl. Acad. Sci. USA, 1997, 94, 10699–10704 CrossRef CAS.
- C. H. Tung and R. Weissleder, Adv. Drug Delivery Rev., 2003, 55, 281–294 CrossRef CAS.
- M. E. Lindgren, M. M. Hallbrink, A. M. Elmquist and U. Langel, Biochem. J., 2004, 377, 69–76 CrossRef CAS.
- M. Lewin, N. Carlesso, C. H. Tung, X. W. Tang, D. Cory, D. T. Scadden and R. Weissleder, Nat. Biotechnol., 2000, 18, 410–414 CrossRef CAS.
- M. Zhao, M. F. Kircher, L. Josephson and R. Weissleder, Bioconjugate Chem., 2002, 13, 840–844 CrossRef CAS.
- P. E. G. Thoren, D. Persson, E. K. Esbjorner, M. Goksor, P. Lincoln and B. Norden, Biochemistry, 2004, 43, 3471–3489 CrossRef CAS.
- A. Joliot and A. Prochiantz, Nat. Cell Biol., 2004, 6, 189–196 CrossRef CAS.
- G. Drin, S. Cottin, E. Blanc, A. R. Rees and J. Temsamani, J. Biol. Chem., 2003, 278, 31192–31201 CrossRef CAS.
- E. Vives, J. Mol. Recognit., 2003, 16, 265–271 CrossRef CAS.
- J. A. Rottingen and J. G. I., Acta Physiol. Scand., 2000, 169, 203–219 CrossRef CAS.
- R. Rudolf, M. Mongillo, R. Rizzuto and T. Pozzan, Nat. Rev. Mol. Cell Biol., 2003, 4, 579–586 CrossRef CAS.
- R. F. Irvine, Nat. Rev. Mol. Cell Biol., 2003, 4, 586–590 CAS.
- M. J. Berridge, M. D. Bootman and H. L. Roderick, Nat. Rev. Mol. Cell Biol., 2003, 4, 517–529 CrossRef CAS.
- S. K. Lee, J. Y. Lee, M. Y. Lee, S. M. Chung and J. H. Chung, Anal. Biochem., 1999, 273, 186–191 CrossRef CAS.
- J. Zhang, R. E. Campbell, A. Y. Ting and R. Y. Tsien, Nat. Rev. Mol. Cell Biol., 2002, 3, 906–918 CrossRef CAS.
- R. P. Haugland, Handbook of Fluorescent Probes and Research Products, Molecular Probes, 2001 Search PubMed.
- F. Divirgilio, T. H. Steinberg and S. C. Silverstein, Cell Calcium, 1990, 11, 57–62 CrossRef CAS.
- H. A. Clark, R. Kopelman, R. Tjalkens and M. A. Philbert, Anal. Chem., 1999, 71, 4837–4843 CrossRef CAS.
- S. N. Orlov, R. Grygorczyk and S. V. Kotelevtsev, Cell Calcium, 2003, 34, 511–515 CrossRef CAS.
- H. A. Clark, S. L. R. Barker, M. Brasuel, M. T. Miller, E. Monson, S. Parus, Z. Y. Shi, A. Song, B. Thorsrud, R. Kopelman, A. Ade, W. Meixner, B. Athey, M. Hoyer, D. Hill, R. Lightle and M. A. Philbert, Sens. Actuators B: Chemical, 1998, 51, 12–16 CrossRef.
- H. Xu, J. W. Aylott, R. Kopelman, T. J. Miller and M. A. Philbert, Anal. Chem., 2001, 73, 4124–4133 CrossRef CAS.
- H. A. Clark, M. Hoyer, M. A. Philbert and R. Kopelman, Anal. Chem., 1999, 71, 4831–4836 CrossRef CAS.
- M. Brasuel, R. Kopelman, T. J. Miller, R. Tjalkens and M. A. Philbert, Anal. Chem., 2001, 73, 2221–2228 CrossRef CAS.
- H. Xu, J. W. Aylott and R. Kopelman, Analyst, 2002, 127, 1471–1477 RSC.
- N. J. Caron, Y. Torrente, G. Camirand, M. Bujold, P. Chapdelaine, K. Leriche, N. Bresolin and J. P. Tremblay, Mol. Ther., 2001, 3, 310–318 CrossRef CAS.
- L. C. Shriver-Lake, W. B. Gammeter, S. S. Bang and M. Pazirandeh, Anal. Chim. Acta, 2002, 470, 71–78 CrossRef CAS.
- W. van Nieuwenhuyzen and B. F. Szuhaj, Lipid-Fett, 1998, 100, 282–291 Search PubMed.
| This journal is © The Royal Society of Chemistry 2005 |