A method for rapid reaction optimisation in continuous-flow microfluidic reactors using online Raman spectroscopic detection
Shee-Ann
Leung†
,
Richard F.
Winkle†
,
Robert C. R.
Wootton
and
Andrew J.
deMello
*
Department of Chemistry, Imperial College London, Exhibition Road, South Kensington, London, UK SW7 2AZ. E-mail: a.demello@imperial.ac.uk; Fax: +44 (0)207 5945834; Tel: +44 (0)207 5945820
First published on 22nd November 2004
Abstract
An extremely rapid tool for continuous flow synthetic process optimisation is described. A microfluidic reaction system operating in continuous flow is used in conjunction with confocal Raman microscopy to afford rapid molecule synthesis and product quantitation. Accordingly, the approach allows for rapid reaction optimisation within a continuous flow system. Specifically, the catalytic oxidation of isopropyl alcohol (IPA) to acetone using tetra-N-propylammonium perruthanate (TPAP)/N-methylmorpholine N-oxide (NMO) in a radial interdigitated micromixer is studied as a model reaction system. The composition of the reaction effluent can be determined with great facility and information relating to catalyst/co-oxidant ratios, catalyst turnovers and reaction endpoints extracted. Specifically, variation of catalyst and co-oxidant volumetric flow rates between 0 and 50 µL min−1 is used to vary reactant concentrations, define reaction residence times and control product conversions between 0 and 100%. The rapid nature of the system allows chemical information to be gathered and utilised on a sub-minute timescale.
Introduction
The optimisation of reactions in fine or bulk chemistry is vital to the economic success of any synthetic route. Every stage of such a process requires some investigation to ensure that the ideal balance between cost and product yield is met. That said, reaction optimisation is normally a time consuming process. Data for each system can only be gathered by the repeated performance of the reaction under varied conditions. In recent years, robotic systems have been employed to provide a degree of automation when carrying out optimisation studies.1 Such systems are typically used to automate and optimise reagent metering and delivery, reaction processing, product detection and product isolation. This approach, although an improvement over multiple human repeats for the optimisation, is still a complex and time-consuming process, since reactions are performed batch-wise and require extended times to go to completion. Further time delays are incurred since detection and analysis of products is most normally performed off-line using techniques such as high-performance liquid chromatography. For example, a recent automated macroscale optimisation system exhibited a typical throughput of 0.35 reactions hour−1 for the formation of an acid chloride from the corresponding carboxylic acid.2 Here, the rate determining step was the analysis time engendered by the use of HPLC, which limited the analytical throughput to approximately 500 reactions per week. Modern developments in the areas of chemometrics and chemoinformatics can provide more ‘intelligent’ solutions to reaction optimisation.3 Here, statistical routines are used to ‘guide’ repeat experiments towards optimum reaction conditions using the minimum number of iterations. This approach allows a more rapid generation of ‘reaction space’ information than intuitive approaches but is still limited by the reactor instrumentation. Even with robotic synthesis and statistical variation protocols the investment of time and resource in each process is considerable.Subsequent to laboratory-scale optimisation of a reaction, ‘scale-up’ of the reaction scheme is required to afford production-scale quantities of material and industrial viability. To accomplish this process the reaction system often has to be radically adapted, since reaction behaviour on the laboratory-scale is rarely mirrored in the system's performance on a fine to bulk scale. Safety procedures often add to large scale-up costs, and can lead to the abandonment of a synthesis if a particular hazard becomes apparent. Microfluidic devices allow access to fine scale chemistry through the parallel repetition of small scale continuous-flow reactions (termed ‘scale-out’). The advantage of this approach when compared to traditional ‘scale-up’ methods lies primarily in the fact that the chemistry involved does not have to be adapted in any way. To move to a bulk scale is simply a case of performing the same process many times in parallel. For example, if a microfluidic reaction system yields 0.5 g of product per hour, 100 identical devices running in parallel in will generate 50 g of product in the same time. Krummradt et al. have demonstrated the application of microfluidic reactors in an established multistage process for the generation of a fine chemical from the exothermic reaction between a carbonyl compound and an organometallic reagent.4
In the majority of studies reporting the use of microfluidic devices for small molecule synthesis detection has been performed offline by collecting samples for later analysis by conventional GC, MS and GC-MS. This is not only time consuming but generates uncertainty in understanding the reaction profile. Specifically, if reactions are not adequately quenched on removal from the reactor any difference in sampling time will cause large discrepancies in assessment of product yields and conversions. In order to alleviate this problem online detection methods are desirable.
Small volume detection within planar chip devices has conventionally been based around optical measurements.5 This is primarily due to the fact that most common substrates (including glass, quartz and polymeric materials) are optically transparent in the visible regions of the electromagnetic spectrum. A cursory survey of the literature demonstrates that by far the most common method for on-chip detection is laser-induced fluorescence (LIF). LIF approaches have proved popular due to their exceptional sensitivity and low mass detection limits.6 However, LIF techniques suffer from significant drawbacks that prohibit their universal use. These include relatively high instrumental costs, the fact that the majority of molecular species do not fluoresce (or are not easily converted to fluorescent species) and the lack of fingerprinting or structural information accessible from time-integrated fluorescence measurements. When applied to the analysis of small organic molecules LIF techniques are even less attractive due the very low proportion of molecules possessing fluorophores with acceptable fluorescence quantum efficiencies. To overcome these limitations we (and others) have recently investigated the use of Raman spectroscopy for high-information content, on-line detection within microfluidic systems.7 Raman spectroscopy utilises monochromatic light, usually in the visible region of the spectrum, and is an established tool for real-time chemical fingerprinting. When using optically transparent substrates (such as glass and quartz) Raman spectroscopy allows for the direct on-line detection of analytes in situ, without need for reaction quenching. In addition, the flat, uniform nature of microchannels and substrate surfaces in chip-based systems reduces scatter dispersion and ensures that scattered photons are collected only from the solution (not from the glass substrate), resulting in excellent sensitivities.8 The ability to both identify and quantify analytes present within a microfluidic is particularly advantageous and obviates the time lag incurred by post-reaction separations (e.g., via gas or liquid chromatography).
Raman spectroscopy has been used for real-time monitoring of macroscale reactions.9 For example, Ampiah-Bonney and Walmsley investigated the acid catalysed esterification of ethanol with acetic acid by insertion of a Raman probe into the reaction mixture. In addition, Lee et al. have reported the use of Raman spectroscopic detection to elucidate the kinetics of imine formation in batch reactors.10 As previously noted, such real-time analysis, when operating within a continuous-flow microfluidic regime, allows reaction conditions to be changed and optimised on short timescales and with minimal user intervention.
In the current study we demonstrate the use of Raman spectroscopic detection for rapid, on-line analysis and reaction condition screening of the catalytic oxidation of isopropyl alcohol to acetone using the tetra-N-propylammonium perruthenate (TPAP)/N-methylmorpholine N-oxide (NMO) system. This system, developed by Griffiths and Ley is a highly popular and efficient route to the selective oxidation of secondary alcohols.11 The overall reaction is shown in Scheme 1.
 | ||
| Scheme 1 The catalytic oxidation of isopropyl alcohol to acetone. | ||
The rapidity of data generation using this approach provides an obvious tool for the optimisation chemist as dependencies can be investigated in near real time. The use of Raman spectroscopy also allows structural information to be inferred without time-consuming separations, relieving the analysis lag often encountered in automated systems. The technique shows clear advantages in speed and flexibility over traditional methods.
Experimental
Microfluidic reactor fabrication
For current experiments microfluidic reaction systems were designed so as to allow for rapid and efficient mixing of reactants whilst allowing operation at relatively high volumetric flow rates. Devices were based on a radial interdigitated mixer design (Fig. 1). The microfluidic devices function by purely diffusive mixing between 16 laminae. Each mixer is fabricated in 3-layers. In the first two layers input flows are directed to two circular bus channels which, in turn, split the flow into 8 identical fluid laminae and deliver reagent streams towards a central mixing chamber. The final layer acts as a cap to enclose channels and as a guide for input and output capillaries. During mixing flow streams are brought together alternately in a radial sense and mixing is achieved via diffusion. | ||
| Fig. 1 Three dimensional schematic of a radial interdigitated mixer. Each mixer is fabricated in 3-layers. In the first two layers input flows are directed to two circular bus channels which, in turn, split the flow into 8 identical fluid laminae and deliver reagent streams towards a central mixing chamber. The final layer acts as a cap to enclose channels and as a guide for input and output capillaries. | ||
Microfluidic devices were made in-house using direct-write laser lithography, wet chemical etching and bonding techniques. Briefly, soda lime glass substrate precoated with a positive photoresist (AZ 1518) and a low reflective chromium layer (Nanofilm, Westlake Village, California) was exposed using a DWL system (DWL2.0, Heidelberg Instruments, Heidelberg, Germany) to transfer the channel design. After the photoresist was developed (Microposit 351, Shipley Europe Ltd., Coventry, UK), channels were etched into the glass substrate using a buffered oxide etching solution (HF–NH4F) and external access holes were drilled. To form enclosed channels, a glass cover plate cleaned with concentrated H2SO4 was thermally bonded in a furnace (Heraeus Instruments GmbH, Hanau, Germany) at 585 °C. Fused silica capillary (0.375 mm od, 0.05 mm id) was cemented to the inlet and outlet holes using a thermally cured two-part epoxy (Araldite 2014). Reagents were purchased from Aldrich (Sigma–Aldrich Ltd, Gillingham, Dorset, UK.) and used as received. Dichloromethane (BDH Chemicals, Poole, Dorset, UK) was dried over molecular sieves (4 Å) for 12 h before use. When required, chips were connected using PTFE tubing (350 µm id, Upchurch Scientific, Oak Harbour, USA) which forms a facile pressure seal with the capillary used. The volume of the reaction chamber was 2.3 µl. The 16 convergent channels each had a width of 150 µm and a depth of 50 µm. Fourier number calculations for the fabricated structures indicate that complete mixing (τ = 1) occurs in the outlet capillary at volumetric flow rates of 900 µl min−1 or less and adequate mixing (τ = 0.1) occurs at flow rates lower than 4500 µl min−1. Reynolds numbers remain lower than 20 for all flow rates employed. This is within the region where the dominant flow pattern is expected to be laminar. These data are displayed graphically in Fig. 2.
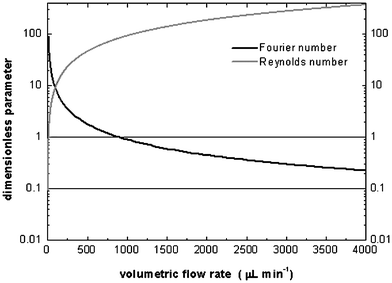 | ||
| Fig. 2 Variation of Reynolds and Fourier numbers as a function of total volumetric flow rate for the radial interdigitated mixer described in Fig. 1. Calculations assume a fluid density of 1 g cm−3, fluid viscosity of 8.91 × 10−4 N s m−2, and a solute diffusion coefficient of 1 × 10−9 m2 s−1. | ||
General method for continuous flow synthesis of acetone from isopropyl alcohol
A solution of tetra-N-propylammonium perruthenate ([nPr4N][RuO4], 40 mg, 0.114 mmol) and N-methylmorpholine-N-oxide (0.27 g, 2.3 mmol) in dichloromethane (1.8 ml) was loaded into one syringe, and a solution of isopropyl alcohol (0.5 ml, 5.9 mmol) in dichloromethane (1.5 ml) into the other. The volumetric flow rate of the TPAP/NMO oxidant mixture was subsequently varied with respect to a constant flow of isopropyl alcohol (25 µl min−1).Raman spectroscopic detection
All Raman spectra were measured using a Jobin–Yvon LabRam Infinity 1010 Microscope. The instrument consisted of an inverted microscope (Olympus System Microscope Model BX40) with 10×/0.25, 50×/0.50 and 100×/0.80 infinity corrected objectives for illumination and scattered light collection. A HeNe laser (17 mW at 633 nm) was used as an excitation source for all experiments. Detection was performed using a CCD array held in a vacuum chamber which was dried and filled with dry nitrogen gas. The CCD detector consisted of an array of 1024 × 256 pixels and 1000 cm−1 is dispersed by the spectrometer over the CCD detector to afford a resolution of 1 cm−1 per pixel. Spectra were acquired using CCD 3000 software (Jobin–Yvon) with a 1 to 60 s integration time and 100 ms rest between acquisitions. An in-house program written in Matlab 6.5 (Mathworks, Cambridge, UK) was used to extract and display data in a three dimensional graphical from, illustrating the effect of a variant versus the Raman spectra obtained.Results
Initial experiments were performed to assess the oxidation of IPA to acetone (with TPAP catalyst and NMO co-oxidant) using a single microfluidic reactor (Fig. 3(a)). Raman spectra were recorded 30 cm downstream of the point of confluence of reagent streams. Complete mixing was expected to occur 3 cm downstream of mixing in all cases, based on calculated Fourier numbers. Reactants were hydrodynamically motivated through the microfluidic mixer, and subsequently through an in-line silica plug (Merck Keiselgel 60, 3 cm long). Raman spectra of stock solutions of acetone and IPA within the microfluidic reactor were recorded and used to generate appropriate calibration curves. For the purposes of the current experiments the isopropyl skeletal stretching bands at 820 cm−1 (IPA) and 780 cm−1 (acetone) are of particular interest since their relative intensities can be used to monitor reaction progress. The average residence times in the reactor were between 1.4 and 4.5 s. | ||
| Fig. 3 (a) Experimental schematic for isopropyl alcohol oxidation where tetra-N-propylammonium perruthenate and N-methylmorpholine-N-oxide are mixed prior to reaction with isopropyl alcohol. (b) Experimental schematic for isopropyl alcohol oxidation where the relative concentration of tetra-N-propylammonium perruthenate and N-methylmorpholine-N-oxide is precisely controlled using a mixer prior to reaction with isopropyl alcohol. | ||
As discussed previously, in this configuration a solution of tetra-N-propylammonium perruthenate (40 mg, 0.114 mmol) and N-methylmorpholine-N-oxide (0.27 g, 2.3 mmol) in dichloromethane was loaded into one syringe, and a solution of isopropyl alcohol (0.5 ml, 5.9 mmol) in dichloromethane in the other. The volumetric flow rate of the TPAP/NMO oxidant mixture was subsequently varied with respect to a constant flow of isopropyl alcohol. As the flow rate of the TPAP/NMO solution was varied between 5 and 50 µl min−1, resulting Raman spectra of the product solutions show significant differences in the relative intensities of the IPA and acetone peaks (at 820 s cm−1 and 780 cm−1, respectively). Fig. 4(a) illustrates the variation of Raman intensity as a function of the TPAP/NMO flow rate and Raman shift. Qualitatively, it can be seen that as the volumetric flow rate of the catalyst/co-oxidant stream is increased the intensity of the IPA isopropyl skeletal stretching band at 820 cm−1 reduces whilst the acetone isopropyl skeletal stretching band at 780 cm−1 increases. From these data, concentration versus TPAP/NMO plots were generated for both reactant and product and are shown in Fig. 4(b). Fig. 4(b) shows that as the flow rate of TPAP/NMO increases so does the amount of IPA converted to acetone. As was expected the sum of the concentrations followed a predictable curve, generated by the dilution effect of increased oxidant flow. This arises because the solvent in which the oxidant is added decreases the overall concentration of IPA and acetone in the detection zone by a known amount. When the flow rate of TPAP/NMO reached 50 µl min−1 the IPA isopropyl skeletal stretching band at 820 cm−1 was indistinguishable from the background, showing that little or no IPA is left unreacted. For our system, this defines the minimum amount of the catalytic solution required for 100% conversion.
 | ||
| Fig. 4 (a) Raman spectra of fluid exiting the microfluidic reactor as a function of TPAP/NMO volumetric flow rate. Raman spectroscopic detection conditions: 4 mW, 632 nm excitation, 15 s integration time and 20 cm−1 resolution. (b) Extracted concentrations of IPA, acetone and total reagent as a function of TPAP/NMO volumetric flow rate. Experimental schematic as shown in Fig. 3(a). | ||
Subsequent studies were performed to assess the effect of catalyst/co-oxidant ratio on conversion of IPA to acetone (i.e., the catalytic turnover of TPAP as a function of the TPAP ∶ NMO ratio). In these experiments, instead of using a pre-mixed TPAP/NMO feedstock, two microfluidic reactors were used in series (Fig. 3(b)). Here, the first reactor is used to mix the catalyst and co-oxidant, the ratio of TPAP to NMO being controlled by variation of TPAP input flow (between 0 and 25 µl min−1). The second reactor is then used (in the same way to initial experiments) to mix the catalyst/co-oxidant stream with isopropyl alcohol. Other experimental parameters were identical with those described previously. Fig. 5(a) illustrates the variation of Raman intensity as a function of the TPAP ∶ NMO ratio and Raman shift. Qualitatively, it can be seen that as the TPAP volumetric flow rate is increased the intensity of the IPA isopropyl skeletal stretching band at 820 cm−1 reduces whilst the acetone isopropyl skeletal stretching band at 780 cm−1 increases. The relationship between the oxidant concentration and the extent of oxidation is shown more clearly when the concentrations of acetone and IPA are plotted against oxidant flow rate for this system (Fig. 5(b)). The plot of reactant concentrations against TPAP solution flow rate indicates that a minimum amount of TPAP is required to achieve 100% conversion; this could be found easily if the TPAP flow rate was increased further but this proved problematic due to solid production (ascribed to lower oxidation state ruthenium species) within the microfluidic channels. Catalytic turnovers for the system were calculated for each volumetric flow rate and shown in Fig. 6. It can be observed that no levelling off of turnovers occurs within the flow regime studies, indicating that catalyst regeneration proceeds smoothly. The maximum number of turnovers measured was 16.8.
 | ||
| Fig. 5 (a) Raman spectra of fluid exiting the microfluidic reactor as a function of TPAP volumetric flow rate. Raman spectroscopic detection conditions: 4 mW, 632 nm excitation, 15 s integration time and 20 cm−1 resolution. (b) Extracted concentrations of IPA, acetone and total reagent as a function of TPAP volumetric flow rate. Experimental schematic as shown in Fig. 3(b). | ||
 | ||
| Fig. 6 Variation of catalytic turnovers as a function of TPAP volumetric flow rate. Experimental schematic as shown in Fig. 3(b). | ||
Conclusions
The combination of continuous flow microfluidic reactors and on-line Raman spectroscopic detection has been demonstrated to yield an efficient and rapid tool for investigating dependencies in solution phase chemical reactions. Specifically, a microfluidic approach was used to transform isopropyl alcohol to acetone using the TPAP/NMO catalytic system under a variety of experimental conditions. Rapid variation in reaction conditions is possible, and the real-time nature of the information gathering system allows serial reaction analysis to be performed an order of magnitude faster than an equivalent parallel reaction system. Significantly, all data in this study were gathered and analysed within the space of 10 min (i.e., experiments could be run at a rate in excess of 0.5 variations min−1). This compares with an estimated timescale of 48 hours when extracting the equivalent from batch, macroscale reactors.The technique has obvious application for reaction optimisation. The limited reaction volumes and rapid rate of information generation represent a significant improvement over existing synthetic systems. High efficiency results can be achieved in a short timescale and for a range of initial reagent/catalyst concentrations. Consequently, the system can be employed to judge the extent of reaction or to gather information about catalyst performance.
Acknowledgements
R.F.W. is grateful to Syngenta and EPSRC for joint funding of a research studentship. S.A.L. is grateful to AstraZeneca for provision of an industrial research studentship. Access to the confocal Raman microscope was kindly provided by Professor Bill Griffiths at Imperial College London.References
- M. Pollard, Org. Proc. Res. Dev., 2001, 5, 273 Search PubMed.
- M. Harre, H. Neh, C. Schultz, U. Tilstam, T. Wessa and H. Weinmann, Org. Proc. Res. Dev., 2001, 5, 335–339 Search PubMed.
- G. E. P. Box, W. G. Hunter and J. S. Hunter, Statistics for Experimenters, Wiley, New York, 1978 Search PubMed; R. Carlson, Design and Optimisation in Organic Synthesis, Elsevier, Amsterdam, 1992 Search PubMed; S. Godbert, Design and Analysis in Chemical Research, ed. R. L. Tranter, Sheffield Academic Press, Sheffield, 2000, ch. 6, pp. 188–236 Search PubMed.
- H. Krummradt, U. Kopp and J. Stoldt, ‘Experiences With the Use of Microreactors in Organic Synthesis’, in Microreaction Technology: 3rd International Conference on Microreaction Technology, Proceedings of IMRET 3, ed. W. Ehrfeld, Springer, Berlin, Germany, 2000, p. 18 Search PubMed.
- M. A. Schwarz and P. C. Hauser, Lab Chip, 2001, 1, 1 RSC.
- A. J. de Mello, Lab Chip, 2003, 3, 29N RSC.
- R. Fortt, R. C. R. Wootton and A. J. de Mello, Micro Total Analysis Systems, 2002, Kluwer Academic Publishers, Dordrecht, 2002, pp. 850–852 Search PubMed; R. Fortt, R. C. R. Wootton and A. J. de Mello, Org. Proc. Res. Dev., 2003, 7, 762–768 Search PubMed; P. D. I. Fletcher, S. J. Haswell and X. Zhang, Electrophoresis, 2003, 24, 3239 Search PubMed; M. Lee, J.-P. Lee, H. Rhee, J. Choo, Y. G. Chai and E. K. Lee, J. Raman Spectrosc., 2003, 34, 737 CrossRef CAS.
- P. A. Walker, M. D. Morris, M. A. Burns and B. N. Johnson, Anal. Chem., 1998, 70, 3766 CrossRef CAS.
- R. J. Ampiah-Bonney and A. D. Walmsley, Analyst, 1999, 124(12), 1817 RSC.
- M. Lee, H. Kim, H. Rhee and J. Choo, Bull. Korean Chem. Soc., 2003, 24, 205 CAS.
- S. V. Ley, Synthesis, 1994, 7, 639 CrossRef.
Footnote |
| † Authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2005 |