Asymmetric cyclopropane synthesis via phosphine oxide mediated cascade reactions
Thomas Boesen, David J. Fox, Warren Galloway, Daniel Sejer Pedersen, Charles R. Tyzack and Stuart Warren*
University Chemical Laboratory, Lensfield Road, Cambridge, UK CB2 1EW. E-mail: sw134@cam.ac.uk
First published on 11th January 2005
Abstract
A silyloxy-THF has been converted into a cyclopropane containing three stereocentres as mixture of diastereoisomers. The mechanism of the reaction has been established and the source of stereochemical leakage proposed. An alternative stereospecific cascade reaction has been discovered.
Cyclopropanes appear in a range of natural products and medicinally active molecules.1 Their asymmetric synthesis has therefore been the subject of much research effort.2 Since the first example of homogeneous metal-catalysed asymmetric synthesis,3,4 many formal additions of carbenes to olefins catalysed by palladium,5 copper,6–8 cobalt and ruthenium,9 and rhodium10 have been reported. Asymmetric versions of the zinc-mediated Simmons–Smith cyclopropanation11–14 have also been used in synthesis.15 Cascade ring-closing reactions involving phosphorus transfer, to generate both nucleophile and leaving group, have yielded cyclopropanes in good yield and, generally, with high stereospecificity and selectivity. Phosphine oxide,16–18 phosphinate19,20 and phosphonium salt21 versions are known.
In previous work we have shown that benzoyloxy phosphine oxides 1 can be lithiated with concomitant benzoyl migration to give ketones 2. In the presence of trimethylsilyl chloride the reaction intermediate is intercepted to give silyloxy-tetrahydrofurans 3.22 These heterocycles have been transformed into cyclopropanes 4via a phosphoryl-transfer ring-closing mechanism (Scheme 1).23,24 We have previously shown that single enantiomers of (benzoyloxy)benzyl-substituted THFs, such as 5 (Scheme 2), can be converted into alkene-diols,25 and it was hoped that these compounds could also be converted stereoselectively into cyclopropanes.
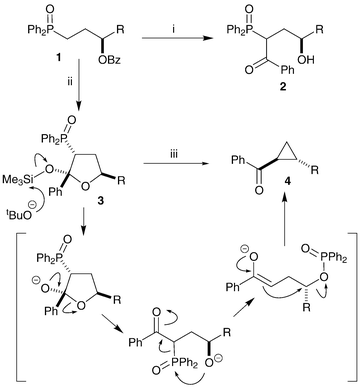 | ||
| Scheme 1 Reagents and conditions: i, LDA, THF, −78 °C; ii, LDA, Me3SiCl, THF, −78 °C; iii, tBuOK, tBuOH. | ||
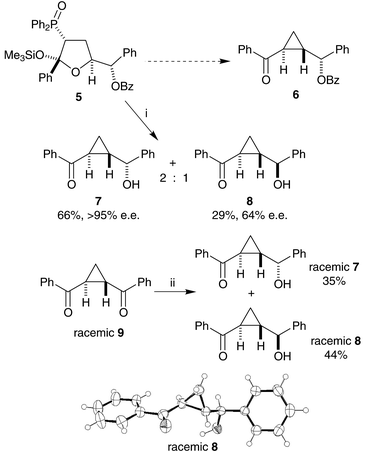 | ||
| Scheme 2 Reagents and conditions: i, tBuOK, tBuOH; ii, NaBH4, MeOH. The X-ray crystal structure of racemic 8 is also shown. | ||
When treated with potassium tert-butoxide in tert-butanol, the conditions previously used to make cyclopropanes (Scheme 2), silyloxy-THF255 did not yield a single stereoisomer of benzoyloxy-cyclopropane 6, but a mixture of two non-benzoylated isomeric cyclopropanes 7 and 8. The two cyclopropanes were both shown to be trans-isomers by reduction of trans-1,2-bisbenzoyl-cyclopropane269 (Scheme 2). Sodium borohydride is mild enough to effect only a mono-reduction of the bis-ketone, but with poor stereoselectivity, to give a mixture of the same cyclopropanes 7 and 8. Racemic compounds 7 and 8 were useful in the accurate calibration of chiral HPLC (Daicel Chiralpak AD column), establishing that the major diastereoisomer formed from THF 5 was as single enantiomer (7), but that the minor diastereoisomer had an enantiomeric excess of only 64% (8). We therefore assumed that the major diastereoisomer formed from THF 5 was the expected one, albeit in de-acylated form. This was confirmed by X-ray crystallography of a racemic sample of the minor cyclopropane diastereoisomer 8 (Scheme 2). The absolute stereochemistry of the minor diastereoisomer was not certain at this stage.
The debenzoylation of the expected product may be involved in the loss of stereochemical information. Consideration of possible reaction intermediates (as neutral compounds), both with benzoyl-protected oxygens (10, 11 and 6) and without (12, 13 and 14), shows that the multistep reaction has many possible pathways (Scheme 3). We hoped that synthesis of some of these possible reaction intermediates, their treatment under the same reaction conditions and analysis of the isolated cyclopropane products would allow for identification of the most significant reaction path and the point of stereochemical scrambling.
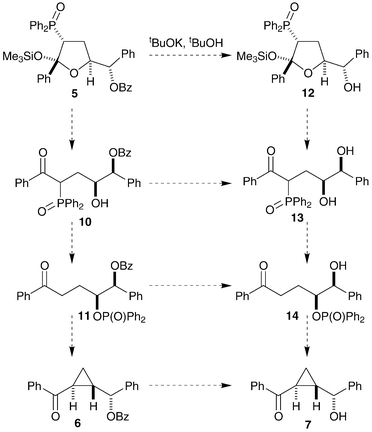 | ||
| Scheme 3 | ||
Hydroxyketone 10 can be made by the opening of THF 5 with TBAF (Scheme 4).25 Keto-phosphinate ester 11 can be selectively synthesised by extended treatment of THF 5 with caesium fluoride in tert-butanol. The different reaction conditions and reaction times in these last two processes show that while the removal of the silyl group and subsequent ring opening are fast reactions, the fluoride-catalysed phosphoryl-transfer from carbon to oxygen (alcohol 10 to phosphinate 11) is relatively slow.17 Diol (4S,5S)-13 was made by cinnamylation and asymmetric dihydroxylation27 of keto-phosphine oxide 16 using AD-mix α (Scheme 4). Cyclopropane 7, obtained from THF 5, was easily benzoylated in high yield to give cyclopropane 6. Compounds 10 and 13 have extremely complicated, uninterpretable NMR spectra, due to the presence of various five- or six-membered cyclic hemi-ketals in solution. The structure of compound 13, as the (4R,5R)-enantiomer, was confirmed by X-ray crystallography (Scheme 4).
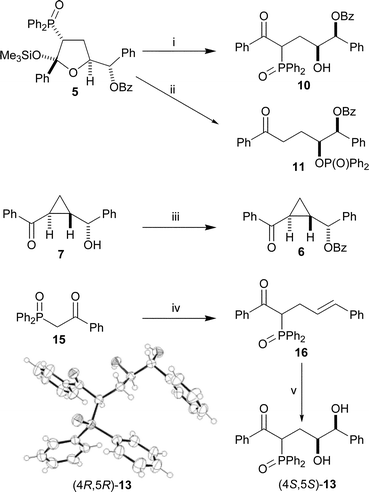 | ||
| Scheme 4 Reagents and conditions: i, TBAF, THF, H2O, 80%; ii, CsF, tBuOH, 80%; iii, PhCOCl, Et3N, DMAP, CH2Cl2, 92%; iv, NaOMe, (E)-cinnamyl bromide, THF, 70%; v, AD-mix α, MeSO2NH2, tBuOH, H2O, 63%. The X-ray crystal structure of (4R,5R)-13, synthesised from 16 using AD-mix-β, is also shown. | ||
Without simple syntheses of debenzoylated THF 12 and phosphinate-alcohol 14, the four available possible reaction intermediates, 10, 11, 13 and 6 as single enantiomers, were separately treated with the reaction conditions used to convert THF 5 into cyclopropanes 7 and 8 (Table 1). All four reactions gave cyclopropane products, and reactions starting with compounds 10, 11 and 13 gave product mixtures similar to that obtained from the reaction of THF 5. The ratio of diastereoisomeric cyclopropanes measured by NMR spectroscopy was roughly 2 to 1 for compounds 7 and 8. In each case the major diastereoisomer 7 was again obtained as a single enantiomer, while the minor diastereoisomer 8 was obtained in 60 to 70% enantiomeric excess, similarly to that obtained from the reaction of THF 5 (64% e.e.). Single enantiomer cyclopropane 6 reacted to give alcohol 7 without loss of stereochemical integrity. These results imply that compounds 10, 11, 13 and THF 5 react to give a common intermediate in the cyclopropanation, which is not, or cannot be made from, cyclopropane 6. Debenzoylated ketone 14 is the only possible common intermediate proposed in Scheme 3 for the reactions producing stereochemical scrambling.
If hydroxyphosphinate ester 14 is the common reaction intermediate in the synthesis of the cyclopropane products 7 and 8 in near-reproducible ratio and e.e., then a mechanism for the synthesis of the three (the single enantiomer of 7 and the two enantiomers of 8), and only three, stereoisomers of cyclopropane can be proposed (Scheme 5). The major diastereoisomer 7 might be formed as a single enantiomer by simple internal displacement of a phosphinate leaving group by the enolate of ketone 14 with inversion of stereochemistry at C4, giving the trans-cyclopropane 7. The basic reaction conditions would also allow the, possibly reversible, oxygen-to-oxygen transfer of the diphenylphosphinoyl group giving hydroxy-phosphinate 17. Base-mediated intramolecular displacement of the phosphinate groups by the oxygens of alcohols 14 and 17 would give a pair of enantiomeric epoxides (2S,3R)-18 and (2R,3S)-18 respectively. Base-mediated ketone enolisation and epoxide-opening cyclopropane formation of each of the enantiomers of intermediate 18 would give the two enantiomers of cyclopropane 8 obtained in the reaction of compounds 5, 10, 11 and 13. This scheme does not permit the synthesis of the other enantiomer of cyclopropane 7. The formation of alcohol 7 requires one inversion at C4, the synthesis of (1R,2R,1′R)-8 involves a net retention of stereochemistry at C4 due to participation of the neighbouring oxygen, and formation of (1S,2S,1′S)-8 involves the inversion of both C4 and C5 in the migration of the epoxide oxygen from C4 to C5. In all cases the final ring closure gives a trans-cyclopropane, independent of the other stereochemistry in the molecule.
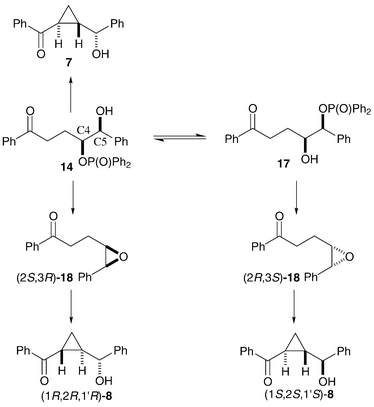 | ||
| Scheme 5 | ||
The proposed epoxide intermediates 18 suggested a route for identification of the major enantiomer of cyclopropane 8 formed from THF 5. Enantiomerically enriched epoxide (2S,3R)-18 was made by manganese-salen catalysed asymmetric epoxidation28 (AE) of cis-olefin2919 (Scheme 6). The sense of asymmetric induction in these oxidation reactions is predictable, and reaction of this isolated epoxide with potassium tert-butoxide gave cyclopropane (1R,2R,1′R)-8. Chiral HPLC analysis of the cyclopropane made via the AE reaction, and comparison with data for the minor diastereoisomeric cyclopropane 8 made from THF 5, identified the (1R,2R,1′R)- isomer as the major enantiomer of cyclopropane 8 produced in the phosphorus-mediated cascade process from THF 5. Epoxide (2S,3R)-18 must therefore form faster than its enantiomer in the cascade reaction.
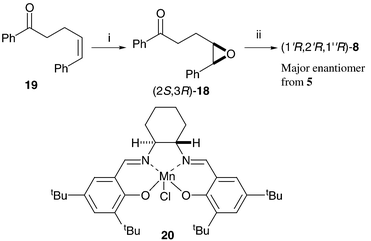 | ||
| Scheme 6 Reagents and conditions: i, NaOCl (aq), 4-PhC5H5NO, 20, CH2Cl2, 67%; ii, tBuOK, tBuOH, 43%. | ||
Epoxide formation can occur only from the free alcohols 14 and 17 so benzoyl ester cleavage is responsible for the loss of stereochemical integrity in the above reactions, and modification of the cyclopropanation reaction conditions is necessary to avoid the ester cleavage. The solution to the problem came unexpectedly during the synthesis of olefin-diols25 from related starting materials. The treatment of bis-benzoate 22 with LDA at −78 °C produced on work-up the acyl transfer product 10 (Scheme 7). Bis-(4-methoxy)benzoate 21 did not react at this temperature, possibly due to the reduction in the electrophilicity of the carbonyls by the electron-donating substituents. We hoped that repetition of the reaction with warming to 0 °C would produce ketone 23, but cyclopropane 24 was the major product of the reaction, as a single stereoisomer and with the para-methoxy benzoate still in place (Scheme 7). Bis-benzoate 22 reacts in a similar way upon warming to give cyclopropane 6, correlating well with the stereochemical assignments made earlier for compounds 7 and 8. The mechanism of formation is probably a similar cascade to that suggested above, but without the unwanted benzoate ester cleavage and subsequent loss of stereochemistry. Compounds 10 and 11 (neutral versions of possible reaction intermediates) were treated with LDA ( −78 to 0 °C) and the same cyclopropane product 6 was produced in both reactions. Removal of the benzoyl group from cyclopropane 6, synthesised directly from compounds 22, 10 and 11, produced alcohol 7 as a single enantiomer, as determined by chiral HPLC. This methodology was shown to work, but with more limited success, on bis-nicotinoyloxy-phosphine oxide 26 (Scheme 8). In this case the basic conditions lead to the cleavage of the more activated acyl group.
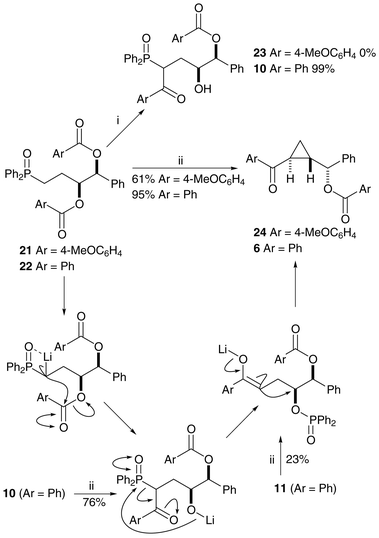 | ||
| Scheme 7 Reagents and conditions: i, LDA, THF, −78 °C; ii, LDA, THF, −78 °C to 0 °C. | ||
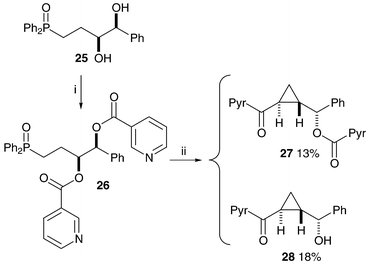 | ||
| Scheme 8 Reagents and conditions: i, 3-Pyr-COCl, DMAP, Et3N, CH2Cl2, 84%; ii, LDA, THF, −78 °C to 0 °C. | ||
In conclusion, the original methodology developed for the synthesis of cyclopropyl ketones from THFs 5 has been shown here to suffer from stereochemical leakage. The problem has been traced to the unexpected cleavage of a benzoate ester by potassium tert-butoxide and the subsequent participation of the resulting hydroxyl group in the adjacent substitution reactions. The proposed mechanism is not only supported by the reactions of possible reaction intermediates, but also by an unusual and reproducible stereochemical divergence where the expected stereoisomer is formed as a single enantiomer, but the other diastereoisomer of product is formed in 60 to 70% e.e. from enantiomerically pure starting material. The formation of a pair of enantiomeric epoxides from the same 1,2-diol is the key to this process, and demonstrates the many roles of the itinerant diphenylphosphinoyl group in this reaction. This problem has however been solved by the discovery of a simpler, more direct cascade reaction for the synthesis of single enantiomers of cyclopropanes with predictable stereochemistry, where the benzoate is not cleaved.
Experimental
All solvents were distilled before use. THF was freshly distilled from a mixture of calcium hydride and lithium aluminium hydride whilst dichloromethane was freshly distilled from calcium hydride. Triphenylmethane was used as an indicator for THF. Methanol was freshly distilled from calcium hydride. Diisopropylamine was dried by stirring over and distilling from calcium hydride (at reduced pressure when necessary) and was stored over activated 4 Å molecular sieves. n-Butyllithium was titrated against diphenylacetic acid before use. All reactions were carried out with oven dried glassware and all reactions in non-aqueous solutions were carried out under an atmosphere of argon.Flash column chromatography was carried out using Merck Kieselgel 60 (230–400 mesh). Thin layer chromatography was carried out on commercially available pre-coated plates (Merck Kieselgel 60F254). Analytical chiral HPLC was performed using a Daicel Chiralpak AD column with a Spectra-Physics SP8800 pump, a Spectra-Physics SP8450 UV detection system and a ChromJet single channel integrator.
Proton, carbon and phosphorus NMR spectra were recorded on Bruker Avance 400 or Avance 500 Fourier Transform spectrometers using an internal deuterium lock. Chemical shifts are quoted in parts per million down field of tetramethylsilane and values of coupling constants (J) are given in Hz. Carbon NMR spectra were recorded with broad band proton decoupling and Attached Proton Test (APT) or DEPT. Complex 1H NMR spectra for molecules containing phosphorus were interpreted with the aid of 1H NMR experiments with phosphorus decoupling.
Melting points were measured on a Stuart Scientific melting point apparatus (SMP 1) and are uncorrected. Infra-red spectra were recorded on a Perkin-Elmer Spectrum One (FT-IR) spectrophotometer. Electron Impact (EI) mass spectra were recorded on a Concept 1H double focusing magnetic sector instrument using a MACH3 data system for high resolution analysis. Fast atom bombardment (FAB) results were obtained from a Kratos MS 890 instrument. Electrospray (ES) mass spectra were recorded using a Micromass Q-Toff instrument and LCMS using a Hewlett Packard HPLC system, eluting with an acetonitrile–water gradient, and in conjunction with positive and negative ion electrospray mass. Microanalyses were carried out by the staff of the University Chemical Laboratory using a CE440 Elemental Analyser from Exeter Analytical, INC.
Optical rotations were recorded on a Perkin Elmer 241 polarimeter (using the sodium D line; 589 nm). Specific rotations are given in units of 10−1 deg dm2 g−1.
(1′S,2′S,1″R)-{2′-[Hydroxy(phenyl)methyl]cyclopropyl}(phenyl)methanone7and (1′S,2′S,1″S)-{2′-[hydroxy(phenyl)methyl]cyclopropyl}(phenyl)methanone8. Potassium tert-butoxide (141 mg, 1.25 mmol) was added to a solution of tetrahydrofuran 5
(269 mg, 0.42 mmol) in tert-butanol (9 cm3) and stirred for 18 h at 35 °C. The mixture was quenched with water (10 cm3) and extracted with dichloromethane (3 × 15 cm3). The combined organic extracts were dried (MgSO4) and evaporated under reduced pressure. The residue was chromatographed (SiO2, hexanes–Et2O, 2 : 1) to give the two diastereoisomeric alcohols7 and 8
(overall 95%). One diastereoisomer (8)
(30 mg, 29%) as white solid; Rf
(hexanes–Et2O, 2 : 1) 0.13; νmax(CH2Cl2)/cm−1 3598 (O–H), 3060 (C–H), 1669 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) and 1603 (CC); δH(400 MHz; CDCl3) 8.10–7.20 (10 H, m, Ph), 4.45 (1 H, d, J 7.0, PhCH), 2.88 (1 H, dt, J 9.0 and 4.5, PhCOCH), 2.06 (1 H, dtd, J 9.0, 7.0 and 4.0, PhCH(OH)CH), 1.90 (1 H, br s, OH), 1.51 (1 H, dt, J 9.0 and 4.5, CHAHB) and 1.19 (1 H, ddd, J 8.5, 6.5 and 4.0, CHAHB); δC(100 MHz; CDCl3) 199.5 (CO), 143.0 (ipso-PhCO), 137.8 (ipso-PhCHOH), 132.9–125.0 (Ph), 76.0 (CHOH), 32.8 (PhCOCH), 23.3 (PhCHOHCH) and 15.6 (CH2); m/z(EI) 234 [14%, (M − H2O)+]
[Found: (M − H2O)+, 234.1043. C17H14O requires M, 234.1032]; e.e. 64%
(Daicel AD column, n-hexane–EtOH, 9 : 1; 1 mL min−1; retention times: major enantiomer, 29 min; minor enantiomer, 21 min). The other diastereoisomer (7)
(70 mg, 66%) as a white solid; Rf(hexanes–Et2O, 2 : 1) 0.08; νmax(CH2Cl2)/cm−1 3599 (O–H), 3062 (C–H), 1670 (CO) and 1605 (CC); δH(400 MHz; CDCl3) 7.90–7.10 (10 H, m, Ph), 4.60 (1 H, d, J 6.0, PhCH), 2.74 (1 H, dt, J 9.0 and 4.5, PhCOCH), 2.05 (1 H, dtd, J 8.5, 6.0 and 4.5 PhCH(OH)CH), 1.95 (1 H, br s, OH), 1.53 (1 H, ddd, J 8.5, 4.5 and 4.0, CHAHB) and 1.26 (1 H, ddd, J 9.0, 6.5 and 4.0 CHAHB); δC(100 MHz; CDCl3) 199.3 (CO), 142.9 (ipso-PhCO), 137.8 (ipso-PhCHOH), 132.8–126.3 (Ph), 74.6 (CHOH), 31.7 (PhCOCH), 22.0 (PhCHOHCH) and 15.2 (CH2); m/z(EI) 252 (10%, M+)
[Found: (M − H2O)+, 234.1033. C17H14O2 requires M, 234.1032]; e.e. >95%
(Daicel AD column, n-hexane–EtOH, 9 : 1; 1 mL min−1; retention times: major enantiomer, 19 min; minor enantiomer, 29 min).
O) and 1603 (CC); δH(400 MHz; CDCl3) 8.10–7.20 (10 H, m, Ph), 4.45 (1 H, d, J 7.0, PhCH), 2.88 (1 H, dt, J 9.0 and 4.5, PhCOCH), 2.06 (1 H, dtd, J 9.0, 7.0 and 4.0, PhCH(OH)CH), 1.90 (1 H, br s, OH), 1.51 (1 H, dt, J 9.0 and 4.5, CHAHB) and 1.19 (1 H, ddd, J 8.5, 6.5 and 4.0, CHAHB); δC(100 MHz; CDCl3) 199.5 (CO), 143.0 (ipso-PhCO), 137.8 (ipso-PhCHOH), 132.9–125.0 (Ph), 76.0 (CHOH), 32.8 (PhCOCH), 23.3 (PhCHOHCH) and 15.6 (CH2); m/z(EI) 234 [14%, (M − H2O)+]
[Found: (M − H2O)+, 234.1043. C17H14O requires M, 234.1032]; e.e. 64%
(Daicel AD column, n-hexane–EtOH, 9 : 1; 1 mL min−1; retention times: major enantiomer, 29 min; minor enantiomer, 21 min). The other diastereoisomer (7)
(70 mg, 66%) as a white solid; Rf(hexanes–Et2O, 2 : 1) 0.08; νmax(CH2Cl2)/cm−1 3599 (O–H), 3062 (C–H), 1670 (CO) and 1605 (CC); δH(400 MHz; CDCl3) 7.90–7.10 (10 H, m, Ph), 4.60 (1 H, d, J 6.0, PhCH), 2.74 (1 H, dt, J 9.0 and 4.5, PhCOCH), 2.05 (1 H, dtd, J 8.5, 6.0 and 4.5 PhCH(OH)CH), 1.95 (1 H, br s, OH), 1.53 (1 H, ddd, J 8.5, 4.5 and 4.0, CHAHB) and 1.26 (1 H, ddd, J 9.0, 6.5 and 4.0 CHAHB); δC(100 MHz; CDCl3) 199.3 (CO), 142.9 (ipso-PhCO), 137.8 (ipso-PhCHOH), 132.8–126.3 (Ph), 74.6 (CHOH), 31.7 (PhCOCH), 22.0 (PhCHOHCH) and 15.2 (CH2); m/z(EI) 252 (10%, M+)
[Found: (M − H2O)+, 234.1033. C17H14O2 requires M, 234.1032]; e.e. >95%
(Daicel AD column, n-hexane–EtOH, 9 : 1; 1 mL min−1; retention times: major enantiomer, 19 min; minor enantiomer, 29 min).
(1′RS,2′RS,1″SR)-{2′-[Hydroxy(phenyl)methyl]cyclopropyl}(phenyl)methanone7and (1′RS,2′RS,1″RS)-{2′-[hydroxy(phenyl)methyl]cyclopropyl}(phenyl)methanone8. Diketone 9 (0.4 g, 1.6 mmol) was dissolved in dry methanol (25 cm3). Sodium borohydride (30 mg, 0.8 mmol) was added and the mixture stirred for 18 hours. Water (10 cm3) was added and the methanol was evaporated under reduced pressure. The aqueous residue was extracted with dichloromethane (3 × 30 cm3), dried (MgSO4) and evaporated under reduced pressure. The residue was chromatographed (SiO2, hexanes–Et2O, 2 : 1) to give after recrystallisation (hexanes–Et2O, 3 : 1) the two diastereoisomeric alcohols7 (176 mg, 44%) and 8 (140 mg, 35%), with analytical data identical to the enantiomerically enriched compounds.
(4S,5S)-5-Benzyloxy-1,5-diphenyl-2-diphenylphosphinoyl-4-hydroxypentan-1-one10. Phosphine oxide 5
(65 mg, 0.10 mmol) was dissolved in tetrahydrofuran (1 cm3) and tetrabutylammonium fluoride (26 mg, 0.10 mmol) was added. The mixture was stirred for 30 min and then quenched with saturated ammonium chloride (0.5 cm3) and the tetrahydrofuran evaporated under reduced pressure. The residue was extracted with ethyl acetate (3 × 2 cm3) and the combined organic extracts were evaporated under reduced pressure. Ethyl acetate (2 cm3) was added and the mixture filtered through a plug of silica. The solvent was evaporated under reduced pressure and the residue was chromatographed (SiO2, EtOAc–hexanes 2 : 1) to give, after recrystallisation (from EtOAc), the keto phosphine oxide10
(46 mg, 80%) as white needles, m.p. 176–178 °C (from EtOAc); Rf(EtOAc–hexanes, 2 : 1) 0.24; [α]23D
−24.0 (c 0.3, CHCl3); νmax(CH2Cl2)/cm−1 1720 (OCO), 1677 (CO), 1598 (CC), 1438 (P–Ph) and 1177 (PO); m/z 574 (6%, M+)
(Found M+, 574.1891. C36H31O5P requires M, 574.1909); anal. (Found: C, 75.0; H, 5.4. C36H31O5P requires C, 75.3; H, 5.4%); the NMR spectroscopic data were complex and inconclusive.
(4S,5S)-5-Benzoyloxy-1,5-diphenyl-4-diphenylphosphinyloxypentan-1-one11. Tetrahydrofuran 5
(0.40 g, 0.62 mmol) was dissolved in tert-butanol (15 cm3) and caesium fluoride (195 mg, 1.24 mmol) added. The mixture was stirred for 48 h at 30 °C, quenched with saturated ammonium chloride (1.5 cm3) and extracted with dichloromethane (3 × 10 cm3). The combined organic extracts were dried (Na2SO4) and evaporated under reduced pressure to yield after recrystallisation (from CH2Cl2) the phosphinate ester11
(283 mg, 80%) as colourless needles, m.p. 71–72 °C (from CH2Cl2); Rf(EtOAc–hexanes 1 : 1) 0.34; [α]24D
−6.0 (c 0.55, CHCl3); νmax(CH2Cl2)/cm−1 1720 [PhCO(O)], 1686 (PhCO), 1598 (CC), 1438 (P–Ph) and 1178 (PO); δH(400 MHz; CDCl3) 7.90–7.85 (2 H, m, Ph), 7.83–7.75 (2 H, m, Ph), 7.70–7.10 (21 H, m, Ph), 6.17 (1 H, d, J 8.0, PhCH), 5.24 (1 H, qd, J 9.0 and 4.0, POCH), 2.98 (2 H, t, J 7.5, PhCOCH2) and 2.10–1.90 (2 H, m, PhCOCH2CH2); δC(100 MHz; CDCl3) 198.6 (PhCOCH2), 165.3 (PhCO2), 136.6 (d, J 54.0, ipso-PPh), 133.8–127.0 (m, Ph2PO and 3 × Ph), 129.4 (ipso-Ph) 78.1 (PhCH), 77.2 (POCHCH2), 34.3 (POCHCH2CH2) and 26.1 (POCHCH2CH2); m/z(ES) 597 (73%, MNa+)
(Found: MNa+, 597.1789. C36H31O5PNa requires M, 597.1807).
1,5-Diphenyl-2-diphenylphosphinoylpent-4-en-1-one16. By the method of Torr and Warren,30 ketone 15
(3.0 g, 9.4 mmol) and sodium methoxide (567 mg, 10.5 mmol) was stirred in tetrahydrofuran (70 cm3) at room temperature under argon for 5 min. A solution of cinnamyl bromide (2.04 g, 10.3 mmol) in tetrahydrofuran (15 cm3) was added and the mixture stirred at room temperature for 19 h. Saturated ammonium chloride solution (20 cm3) was added, followed by water (20 cm3). The tetrahydrofuran was evaporated under reduced pressure and the aqueous residue was extracted with ethyl acetate (3 × 100 cm3). The combined organic extracts were dried (Na2SO4) and evaporated under reduced pressure to give a white solid which, on recrystallisation (from ethyl acetate), gave the alkylated phosphine oxide16
(2.84 g, 70%) as white needles, m.p. 153–155 °C (from EtOAc–hexanes, 2 : 1); Rf(EtOAc) 0.42; νmax(CH2Cl2)/cm−1 1676 (CO) and 1597 (CC); δH(400 MHz; CDCl3) 7.95–7.88 (2 H, m, Ph), 7.81–7.67 (4 H, m, Ph), 7.52–7.07 (14 H, m, Ph), 6.32 (1 H, d, J 16.0, PhCHCH), 6.02 (1 H, dt, J 17.0 and 7.0, PhCHCH), 4.68 (1 H, ddd, J 12.5, 11.0 and 7.5, PCH), 3.20–3.07 (1 H, m, CHAHB) and 2.89–2.78 (1 H, m, CHACHB); δC(100 MHz; CDCl3) 197.4 (CO), 138.1 (ipso-PhCO), 136.9 (ipso-PhCHCH), 132.9 (d, J 63.0, ipso-PhP), 132.5–126 (PhP, PhCO and Ph), 52.2 (d, J 55.0, PCH) and 31.8 (PCHCH2); m/z(EI) 436 (8%, M+) and 201 (100, Ph2PO)
(Found: M+, 436.1612. C29H25O2P requires M, 436.1592).
(4S,5S)-4,5-Dihydroxy-1,5-diphenyl-2-diphenylphosphinoylpentan-1-one13. By the method of Sharpless et al.,27 AD-mix α
(4.82 g) and methanesulfonamide (327 mg, 3.40 mmol) were stirred in 1 : 1 tert-butanol–water (34 cm3) at 25 °C. The solution was cooled to 0 °C whereupon some of the dissolved salts precipitated. Phosphine oxide 16
(1.50 g, 3.40 mmol) was added and the slurry was stirred vigorously for 8 days at 0 °C. Sodium sulfite (5.04 g, 40.0 mmol) was added, the mixture warmed to 20 °C and the resultant slurry stirred for a further 30 min. Ethyl acetate (100 cm3) was added to the reaction mixture, and the aqueous layer was extracted with ethyl acetate (3 × 100 cm3). The combined organic extracts were washed with sodium hydroxide solution (2 × 50 cm3, 2 mol dm−3), brine (80 cm3) and then dried (Na2SO4) and evaporated under reduced pressure to give a crude residue. The residue was chromatographed (SiO2, EtOAc–hexanes, 1 : 1) and then recrystallised (from EtOAc) to give the diol13
(1.0 g, 63%) as white needles, m.p. 177–178 °C (from EtOAc); Rf(EtOAc) 0.28; [α]23D
+28.2 (c 0.55, CHCl3); νmax(CH2Cl2)/cm−1 3300 (O–H), 1677 (CO), 1595 (CC) and 1438 (P–Ph); m/z(EI) 493 (100%, MNa+)
(Found: MNa+, 493.1552. C29H27O4PNa requires M, 493.1545); anal. (Found: C, 73.9; H, 5.8. C29H27O4P requires C, 74.0; H, 5.8%). The NMR spectroscopic data were complex and inconclusive. The enantiomer (4R,5R)-13 was prepared in a similar way using AD mix β and a sample suitable for X-ray crystallography was recrystallised from EtOAc–hexanes.
(1′S,2′S,1″R)-{2-[Benzoyloxy(phenyl)methyl]cyclopropyl}(phenyl)methanone6. Triethylamine (55 mg, 0.54 mmol) and benzoyl chloride (76 mg, 0.54 mmol) were added dropwise to a stirred solution of the hydroxy cyclopropyl ketone 7
(68 mg, 0.27 mmol) and dimethylaminopyridine (22 mg, 0.18 mmol) in anhydrous dichloromethane (2.5 cm3) at ambient temperature. The reaction was stirred for 5 h, quenched with water (5 cm3) and extracted with dichloromethane (3 × 25 cm3). The combined organic extracts were washed with hydrochloric acid (1.5 cm3, 1 mol dm−3), brine (40 cm3) and then dried (Na2SO4), and evaporated under reduced pressure to give a crude residue. The residue was chromatographed (SiO2, Et2O–hexanes, 1 : 2) and then recrystallised (from EtOAc) to give the cyclopropyl ketone6
(88 mg, 92%) as white needles, m.p. 141–143 °C (from EtOAc); Rf(EtOAc) 0.71; [α]23D
+9.33 (c 0.375, CHCl3); νmax(CH2Cl2)/cm−1 1718 (OCO) and 1670 (CO); δH(400 MHz; CDCl3) 8.12 (2 H, dd, J 8.5 and 1.5, ortho-PhCO), 7.81 (2 H, dd, J 8.5 and 1.5, ortho-PhCO2), 7.60–7.19 (11 H, m, 3 × Ph), 5.94 (1 H, d, J 7.0, PhCH), 2.76 [1 H, dt, J 8.5 and 5.0, PhC(O)CH], 2.30 (1 H, dddd, J 8.5, 7.0, 6.5 and 4.0, PhCHCH), 1.64 (1 H, ddd, J 9.0, 5.0 and 4.0, CHACHB) and 1.39 (1 H, dt, J 8.5, 6.5 and 4.0, CHACHB); δC(100 MHz; CDCl3) 198.6 (CO), 165.7 (OCO), 139.0 (ipso-PhCO2), 137.6 (ipso-PhCO), 133.2–126.7 (PhCO2, PhCO and Ph), 130.1 (ipso-Ph), 62.7 (CHO), 29.5 [PhC(O)CH], 22.4 (PhCHCH) and 15.9 (CH2); m/z(EI) 356 (38%, M+) and 251 (50, M+
− PhCO)
(Found M, 356.1411. C12H20O3 requires M, 356.1412); anal. (Found: C, 80.6; H, 5.7. C12H20O3 requires C, 80.8; H, 5.7%); e.e. >95%
(n-hexane–EtOH, 9 : 1). A sample was debenzoylated with potassium tert-butoxide in tert-butanol to give the benzyl alcohol 7 with e.e. >95%
(n-hexane–EtOH, 9 : 1).
(1′S,2′S,1″R)-[2′-{Hydroxy(phenyl)methyl}cyclopropyl](phenyl)methanone7. Potassium tert-butoxide (76 mg, 0.68 mmol) and cyclopropyl ketone 6 (81 mg, 0.23 mmol) were dissolved in tert-butanol (5 cm3) and stirred for 1 h at 35 °C. The mixture was quenched with saturated ammonium chloride (2 cm3) and water (1 cm3) was added. The aqueous layer was extracted with dichloromethane (3 × 10 cm3). The combined organic extracts were dried (Na2SO4) and evaporated under reduced pressure. The residue was chromatographed (SiO2, Et2O–hexanes, 1 : 2) to give the alcohol7 (55 mg, 96%) which was spectroscopically identical to that reported above. e.e. >95% (n-hexane–EtOH, 9 : 1).
Synthesis of cyclopropanes7and8from keto-phosphine oxide13. Potassium tert-butoxide (71 mg, 0.64 mmol) and diol 13 (100 mg, 0.213 mmol) were dissolved in tert-butanol (6 cm3) and stirred for 18 h at 35 °C. The mixture was quenched with water (2 cm3) and extracted with dichloromethane (3 × 10 cm3). The combined organic extracts were dried (Na2SO4) and evaporated under reduced pressure. The residue was chromatographed (SiO2, hexanes–Et2O 3 : 1) to give the two diastereoisomeric alcohols7 (30 mg, 55%) and 8 (16 mg, 30%), which were spectroscopically identical to those above. The major diastereoisomer 7 has an e.e. >95% (n-hexane–EtOH, 9 : 1). The minor diastereoisomer 8 has an e.e. of 60% (n-hexane–EtOH, 9 : 1).
Synthesis of cyclopropanes7and8from keto-phosphine oxide10. Potassium tert-butoxide (58 mg, 0.52 mmol) and mono-benzoate 10 (0.10 g, 0.17 mmol) were dissolved in tert-butanol (4.8 cm3) and stirred for 18 h at 35 °C. The mixture was quenched with water (2 cm3) and extracted with dichloromethane (3 × 10 cm3). The combined organic extracts were dried (Na2SO4) and evaporated under reduced pressure. The residue was chromatographed (SiO2, hexanes–Et2O 3 : 1) to give the two diastereoisomeric alcohols7 (11 mg, 26%) and 8 (5 mg, 12%), which were spectroscopically identical to those reported above. The major diastereoisomer 7 has an e.e. >95% (n-hexane–EtOH, 9 : 1). The minor diastereoisomer 8 has an e.e. of 70% (n-hexane–EtOH, 9 : 1).
Synthesis of cyclopropanes7and8from keto-phosphine oxide11. Potassium tert-butoxide (58 mg, 0.52 mmol) and the phosphinate ester 11 (100 mg, 0.17 mmol) were dissolved in tert-butanol (2 cm3) and stirred for 18 h at 35 °C. The mixture was quenched with water (1 cm3) and extracted with dichloromethane (3 × 10 cm3). The combined organic extracts were dried (Na2SO4) and evaporated under reduced pressure. The residue was chromatographed (SiO2, hexanes–Et2O 2 :1) to give the two diastereoisomeric alcohols7 (17 mg, 39%) and 8 (12 mg, 27%), which were spectroscopically identical to those reported above. The major diastereoisomer 7 has an e.e. >95% (n-hexane–EtOH, 9 : 1). The minor diastereoisomer 8 has an e.e. of 66% (n-hexane–EtOH, 9 : 1).
(4S,5S)-5-Benzyloxy-1,5-diphenyl-2-diphenylphosphinoyl-4-hydroxypentan-1-one10. A solution of LDA was prepared by the dropwise addition of n-butyllithium (0.60 cm3 of a 2.0 mol dm−3 solution in hexanes, 1.2 mmol) to a stirred solution of diisopropylamine (121 mg, 1.20 mmol) in dry tetrahydrofuran (10 cm3) at −78 °C. The prepared LDA was added dropwise to a solution of benzoate ester 21 (575 mg, 1 mmol) in dry tetrahydrofuran (10 cm3) at −78 °C. The reaction mixture was stirred at −78 °C for 18 h and quenched with saturated ammonium chloride (2 cm3). The tetrahydrofuran was evaporated under reduced pressure and the residue was extracted with ethyl acetate (3 × 15 cm3). The organic layer was washed with brine (10 cm3), dried (Na2SO4) and evaporated under reduced pressure to give after recrystallisation (from EtOAc) the mono-benzoate ketone10 (570 mg, 99%) as white needles which was spectroscopically identical to that reported above.
(2S,3R)-2-Phenyl-3-(3′-oxo-3′-phenylpropyl)oxirane18. Ketone2919
(70 mg, 0.3 mmol), 4-phenylpyridine-N-oxide (15 mg, 89µmol), and catalyst (S,S)-20 were suspended in CH2Cl2
(2 cm3) and cooled to 4 °C with stirring. To the cooled solution aqueous NaOCl (2 cm3) was added and the slurry stirred vigorously at 4 °C for 24 hours. The reaction mixture was transferred to a separation funnel with brine (20 cm3) and extracted with CH2Cl2
(2 × 20 cm3). The combined organic phases were dried (Na2SO4), filtered and concentrated in vacuo to give a brown gum that was purified by Dry Column Vacuum Chromatography31
(id 4 cm; 20 cm3 fractions; 4 × hexanes; 2.5–25% EtOAc in hexanes (v/v), 2.5% increments; two fractions of each solvent mixture) to give epoxide17
(51 mg, 67%) as white needles; m.p. (EtOAc, hexanes) 141–143 °C; [α]23D
+12.2 (c 1, CHCl3); νmax(CHCl3)/cm−1 1681 (CO), and 1227 (C–O); δH
(500 MHz; CDCl3)
δ 7.87 (2 H, d, J 7.2, ortho-PhCO), 7.49 (1 H, t, J 7.7, para-PhCO), 7.43 (2 H, t, J 7.7, meta-PhCO), 7.37–7.28 (5 H, m, Ph), 4.14 (1 H, d, J 4.2, PhCH), 3.37 (1 H, ddd, J 7.3, 5.3 and 4.3, PhCHCH), 3.05 (2 H, t, J 7.3, CH2CO), 1.80 (1 H, dtd, J 14.3, 7.6 and 5.6, CH2CH2CO), and 1.73 (1 H, dq, J 14.4 and 7.2, CH2CH2CO); δC
(126 MHz; CDCl3)
δ 199.0 (CO), 136.6 (ipso-Ph), 135.2 (ipso-Ph), 133.1 (para-PhCO), 128.59, 128.58, 128.2, 128.0, 126.4 (Ph), 58.6 (CHCH2), 57.7 (CH2CO), 34.9 (PhCH) and 21.9 (CH2CH); m/z(ESI+) found 275.1039 (C17H16O2Na requires 275.1048).
(1′R,2′R,1″R)-{2′-[Hydroxy(phenyl)methyl]cyclopropyl}(phenyl)methanone8. Epoxide 18 (37 mg, 0.15 mmol) was dissolved in tert-butanol (4.5 cm3) and heated to 35 °C. Potassium tert-butoxide (54 mg, 0.48 mmol) was added and the reaction stirred at 35 °C for 24 hours. The reaction mixture was transferred to a separation funnel with half saturated brine (20 cm3) and extracted with ethyl acetate (3 × 10 cm3). The combined organic phases were dried (Na2SO4), filtered and concentrated in vacuo to give a yellow solid that was purified by Dry Column Vacuum Chromatography31 (id 4 cm; 20 cm3 fractions; 4 × hexanes; 5–75% EtOAc in hexanes (v/v), 5% increments) to give cyclopropane (1′R,2′R,1″R)-8 (16 mg, 43%) as a white solid that was spectroscopically identical to that reported above; e.e. >90% (n-hexane–EtOH, 9 : 1, v/v).
(1′S,2′S,1″R)-{2-[4-Methoxybenzoyloxy(phenyl)methyl]cyclopropyl}(4-methoxyphenyl)methanone24: n-Butyllithium (2 mmol, 2.5 M in hexanes) was added to a solution of diisopropylamine (202 mg, 2 mmol) in THF (5 cm3) at 0 °C and stirred for 10 minutes. The solution was then added to (1S,2S)-bis(4-methoxybenzoyloxy)-4-diphenylphosphinoyl-1-phenylbutane 21
(634 mg, 1 mmol) in THF at −78 °C and stirred for 12 h at the same temperature and then 12 h at 0 °C. The reaction was partitioned between water and dichloromethane, and the organic layer was dried (Na2SO4), evaporated and the residue was chromatographed (SiO2, Et2O–hexanes, 1 : 2) to give cyclopropane 24 as an oil (254 mg, 61%); νmax(CH2Cl2)/cm−1 1707 (OCO), 1660 (CO) and 1605 (CC); δH(400 MHz; CDCl3) 8.05 (2 H, d, J 9, ortho-MeOC6H4CO), 7.87 (2 H, d, J 9, ortho-MeOC6H4CO), 7.48 (2 H, d, J 7, ortho-Ph), 7.35 (2 H, t, J 7.5, meta-Ph), 7.29 (1 H, tt, J 7.5 and 2.5, para-Ph), 6.93 (2 H, d, J 9, meta-MeOC6H4CO), 6.88 (2 H, d, J 9, meta-MeOC6H4CO), 5.90 (1 H, d, J 7.0, PhCH), 3.84 (3 H, s, MeO), 3.83 (3 H, s, MeO), 2.70 (1 H, dt, J 9 and 4.5, PhCOCH), 2.23 (1 H, dddd, J 8.5, 7.0, 6.5 and 4.0, PhCHCH), 1.58 (1 H, ddd, J 9.0, 5.0 and 4.0, CHACHB) and 1.39 (1 H, ddd, J 9.0, 6.0 and 4.0, CHACHB); δC(100 MHz; CDCl3) 197.4 (CO), 165.9, 164.0, 163.8 (CO2 and MeOC
× 2), 139.0 (ipso-Ph), 137.6 (ipso-MeC6H4CO), 131.0 (ipso-Ph), 128.7 (para-Ph), 132.2, 130.7, 129.0, 127.2 (ortho-MeC6H4CO × 2 and ortho- and meta-Ph), 114.1 (meta-MeC6H4CO × 4), 77.0 (CHO), 55.8 (CH3
× 2) 29.6, 22.4 (PhCHCH and ArCOCH) and 15.8 (CH2); m/z(EI) 416.16391 (C26H24O5 requires 416.16238).
Synthesis of cyclopropane6from benzoate ester22. A solution of LDA was prepared by the dropwise addition of n-butyllithium (0.23 cm3 of a 1.7 mol dm−3 solution in hexanes, 0.39 mmol) to a stirred solution of diisopropylamine (40 mg, 0.39 mmol) in dry tetrahydrofuran (2 cm3) at −78 °C. The prepared LDA was added dropwise to a solution of benzoate ester 22 (205 mg, 0.36 mmol) in dry tetrahydrofuran (2 cm3) at −78 °C. The reaction mixture was stirred at −78 °C for 13 h, warmed to 0 °C and stirred for a further 27 h. The reaction was quenched with saturated ammonium chloride (1 cm3) and the tetrahydrofuran evaporated under reduced pressure. The residue was extracted with dichloromethane (3 × 10 cm3). The combined organic extracts were dried (Na2SO4) and evaporated under reduced pressure. The residue was chromatographed (SiO2, Et2O–hexanes, 1 : 2) and then recrystallised (from EtOAc) to give the cyclopropyl ketone6 (121 mg, 95%) which was spectroscopically identical to that reported above. A sample was debenzoylated with potassium tert-butoxide in tert-butanol, as above, to give the benzyl alcohol 7 with e.e. >95% (n-hexane–EtOH, 9 : 1).
Synthesis of cyclopropane6from keto-phosphine oxide10. A solution of LDA was prepared by the dropwise addition of n-butyllithium (0.04 cm3 of a 1.7 mol dm−3 solution in hexane, 68 µmol) to a stirred solution of diisopropylamine (6 mg, 0.06 mmol) in dry tetrahydrofuran (0.5 cm3) at −78 °C. The prepared LDA was added dropwise to a solution of phosphine oxide 10 (32 mg, 55.7 µmol) in dry tetrahydrofuran (0.5 cm3) at −78 °C. The reaction mixture was stirred for 11 h, warmed to 0 °C and stirred for a further 27 h. The reaction was quenched with saturated ammonium chloride (0.5 cm3) and the tetrahydrofuran was evaporated under reduced pressure and the residue was extracted with ethyl acetate (3 × 5 cm3). The combined organic extracts were dried (Na2SO4) and evaporated under reduced pressure. The residue was chromatographed (SiO2, hexanes–EtOAc, 2 : 1) to give the starting material (4 mg, 23%) and, after recrystallisation (from EtOAc), cyclopropyl ketone6 (15 mg, 76%) as white needles, which was spectroscopically identical to that reported above.
Synthesis of cyclopropane6from phosphinate oxide11. A solution of LDA was prepared by the dropwise addition of n-butyllithium (0.04 cm3 of a 2.7 mol dm−3 solution in hexanes, 96 µmol) to a stirred solution of diisopropylamine (10 mg, 96 µmol) in dry tetrahydrofuran (2 cm3) at −78 °C. The prepared LDA was added dropwise to a solution of phosphinate ester 11 (50 mg, 0.09 mmol) in dry tetrahydrofuran (2 cm3) at −78 °C. The reaction mixture was stirred at −78 °C for 11 h, warmed to 0 °C and stirred for a further 27 h. The reaction was quenched with saturated ammonium chloride (1 cm3) and the tetrahydrofuran evaporated under reduced pressure. The residue was extracted with dichloromethane (3 × 10 cm3). The combined organic extracts were dried (Na2SO4) and the organic layer evaporated under reduced pressure. The residue was chromatographed (SiO2, hexanes–Et2O, 2 : 1) to give, after recrystallisation (from EtOAc), the cyclopropyl ketone6 (7 mg, 23%) as white needles, which was spectroscopically identical to that reported above.
(1S,2S)-1,2-Bis(3-nicotinoyloxy)-4-diphenylphosphinoyl-1-phenylbutane26. Triethylamine (96.0 mg, 0.95 mmol) was added dropwise to a stirred solution of diol2525
(71 mg, 0.19 mmol) and dimethylaminopyridine (43 mg, 0.35 mmol) in dry dichloromethane (2 cm3) at room temperature. Nicotinoyl chloride hydrochloride (170 mg, 0.95 mmol) was added and the reaction was stirred for 18 h. The reaction was quenched with water (0.5 cm3) and extracted with dichloromethane (3 × 10 cm3). The combined organic extracts were dried (Na2SO4) and evaporated under reduced pressure. The residue was chromatographed (SiO2, EtOAc–MeOH, 19 : 1) to give, after recrystallisation (from EtOAc–hexanes, 9:1), the bis-nicotinoyl phosphine oxide26
(97 mg, 84%) as white needles, m.p. 166–168 °C (from EtOAc–hexanes, 9 : 1); Rf(EtOAc–MeOH, 19 : 1) 0.18; [α]23D
+7.9 (c 1.55, CHCl3); νmax(CH2Cl2)/cm−1 1731 (CO), 1592 (CC) and 1438 (P–Ph); δH(400 MHz; CDCl3) 9.12 (1 H, d, J 1.5, NCHCCO), 9.10 (1 H, d, J 1.5, NCHCCO), 8.72 (1 H, dd, J 5.0 and 1.5, NCHCH), 8.69 (1 H, dd, J 5.0 and 1.5, NCHCH), 8.19 (1 H, dt, J 8.0 and 2.0, NCHCHCH), 8.15 (1 H, dt, J 8.0 and 2.0, NCHCHCH), 7.66–7.56 (4 H, m, ortho-PPh), 7.52–7.27 (13 H, m, PPh, Ph and 2 × NCHCH), 6.13 (1 H, d, J 7.5, PhCH), 5.75 (1 H, td, J 7.0 and 6.0, PhCHCH), 2.40–2.20 (2 H, m, PCH2) and 2.05–1.91 (2 H, m, PCH2CH2); δC(100 MHz; CDCl3) 164.6, 164.0 (2 × CO), 153.8, 153.7, 150.8, 137.1, 137.0 (Py), 135.4 (ipso-Ph), 132.1 (d, J 99.6, ipso-PPh), 132.0 (d, J 99.6, ipso-PPh), 132.0 (d, J 2.5, para-PPh), 131.9 (d, J 2.5, para-PPh), 130.7 (d, J 9.0, ortho-PPh), 130.6 (d, J 9.0, ortho-PPh), 129.2 (para-CPh), 129.0 (ortho-CPh), 128.8 (d, J 12.0, meta-PPh), 128.7 (d, J 11.5, meta-PPh), 127.3 (meta-CPh), 125.4 (ipso-Py), 125.3 (ipso-Py), 123.4, 123.3 (Py), 77.1 (PhCH), 75.7 (d, J 15.0, PCH2CH2CH), 25.6 (d, J 72.0, PCH2) and 23.1 (PCH2CH2); m/z(ES) 599 (100%, MNa+)
(Found MNa+, 599.1729. C34H29O5N2PNa requires M, 599.1712); anal. (Found: C, 70.7; H, 5.1; N, 4.9. C30H29O5PN2 requires C, 70.8; H, 5.1; N 4.9%).
(1′S,2′S,1″R)-[2′-{3-Nicotinoyloxy(phenyl)methyl}cyclopropyl](3-nicotinoyl)methanone27and (1′S,2′S,1″R)-[2′-{hydroxy(phenyl)methyl}cyclopropyl](3-nicotinoyl)methanone28. A solution of LDA was prepared by the dropwise addition of n-butyllithium (0.32 cm3 of a 1.7 mol dm−3 solution in hexanes, 0.54 mmol) to a stirred solution of diisopropylamine (55 mg, 0.54 mmol) in dry tetrahydrofuran (2.5 cm3) at −78 °C. The prepared LDA was added dropwise to a stirred solution of phosphine oxide 26
(285 mg, 0.50 mmol) in dry tetrahydrofuran (2.5 cm3) at −78 °C. The reaction mixture was stirred for 11 h, subsequently warmed to 0 °C and stirred at 0 °C for an additional 27 h. The reaction was quenched with saturated ammonium chloride (1 cm3) and the tetrahydrofuran was evaporated under reduced pressure. The residue was extracted with ethyl acetate (3 × 5 cm3). The combined organic extracts were dried (Na2SO4) and evaporated under reduced pressure. The residue was chromatographed (SiO2, EtOAc–hexanes, 1 : 2) to give the cyclopropane27
(24 mg, 13%) as a white gum and the hydroxy-cyclopropane28
(23 mg, 18%) as a colourless oil. The cyclopropane27 had the following data: Rf(EtOAc–hexanes, 2 : 1) 0.21; [α]23D
+7.6 (c 1.2, CHCl3); νmax(CH2Cl2) 3039 (C–H) 1723 (OCO), 1676 (PyCO) and 1590 (CC); δH(400 MHz; CDCl3) 9.27 (1 H, br s, Py-C2), 9.06 (1 H, br s, Py-C2), 8.79 (1 H, br s, Py-C6), 8.75 (1 H, br s, Py-C6), 8.32 (1H, dt, J 8.0 and 2.0, Py-C4), 8.07 (1 H, dt, J 8.0 and 2.0, Py-C4), 7.49–7.31 (7 H, m, Ar), 5.90 (1 H, d, J 7.5, PhCH), 2.69 [1 H, dt, J 8.0 and 4.5, PyC(O)CH], 2.33 (1 H, dddd, J 8.5, 7.5, 6.5 and 4.0, PhCHCH), 1.68 (1 H, dt, J 9.0 and 4.5, CHACHB) and 1.42 (1 H, ddd, 8.0, 6.5 and 4.0, CHACHB); δC(125 MHz; CDCl3) 197.3 (CHCO), 164.4 (CO2), 153.7, 153.4, 150.9, 149.4 (Py), 138.2 (ipso-Ph), 137.2, 135.3 (Py) 132.7 (ipso-Py), 128.9 (ortho-Ph), 128.8 (para-Ph), 126.8 (meta-Ph), 126.2 (ipso-Py), 123.6, 123.4 (Py), 77.7 (PhCH), 29.9 (CHCO), 22.6 (PhCHCH) and 16.6 (CH2); m/z(EI) 357 [6%, (M − H)+]
[Found (M − H)+, 357.1231. C22H17O3N2 requires M, 357.1239]. The hydroxy cyclopropane28 had the following data: Rf(EtOAc–hexane, 2 : 1) 0.18; [α]23D
+4.8 (c 1.1, CHCl3); νmax(CH2Cl2) 3688 (O–H), 3048 (C–H), 1673 (PyCO) and 1602 (CC); δH(400 MHz; CDCl3) 9.34–9.20 (1 H, br m, Py-C2), 8.87–8.73 (1 H, br m, Py-C2), 8.28 (1 H, br d, J 7.5, Py-C4), 7.57–7.47 (1 H, br m, Py-C5), 7.43–7.27 (5 H, m, Ph), 4.73 (1 H, d, J 5.5, PhCH), 2.75 [1 H, dt, J 8.0 and 4.5, C(O)CH], 2.11 (1 H, J 8.0, 6.5, 5.5 and 4.0, PhCHCH), 1.61 (1 H, ddd, J 8.5, 4.5 and 4.0, CHACHB) and 1.41 (1 H, ddd, J 8.0, 6.5 and 3.5, CHACHB); δC(125 MHz; CDCl3) 197.9 (CO), 152.5 (Py), 149.0, 142.5 (ipso-Ph), 128.7 (ortho-Ph), 128.2 (para-Ph), 126.2 (meta-Ph), 126.0 (ipso-Py), 123.9 (Py), 73.7 (CHOH), 32.9 (PyCOCH), 22.3 (PhCHOHCH) and 15.5 (CH2); m/z(EI) 252 [8%, (M − H)+]
[Found (M − H)+, 252.1020. C16H14O2N requires M, 252.1024].
Crystal data for 8: C17H16O2, M = 252.30, orthorhombic, space group = Pccn, a = 13.1650(8), b = 28.102(2), c = 7.2735(3) Å, U = 2690.9(3) Å3, Z = 8, µ(Mo-Kα) = 0.080 mm−1, 8455 reflections measured at 180(2) K using an Oxford Cryosystems Cryostream cooling apparatus, 1731 unique (Rint = 0.044); R1 = 0.058, wR2 = 0.118 [I > 2σ(I)]. The structure was solved with SHELXS-97,32 and refined with SHELXL-97.32
Crystal data for 13: C29H27O4P, M = 470.48, orthorhombic, space group = P2(1)2(1)2(1), a = 10.7399(6), b = 12.8444(8), c = 17.6182(11) Å, U = 2430.4(3) Å3, Z = 4, µ(Mo-Kα) = 0.147 mm−1, 14107 reflections measured at 180(2) K using an Oxford Cryosystems Cryostream cooling apparatus, 4251 unique (Rint = 0.106); R1 = 0. 069, wR2 = 0.120 [I > 2σ(I)]. The absolute structure parameter (Flack parameter) 0.2(4) indicates that the absolute structure has been assigned correctly, and this is in agreement with the expected configuration. The structure was solved with SHELXS-97,32 and refined with SHELXL-97.32
CCDC reference numbers 249431 and 249432. See http://www.rsc.org/suppdata/ob/b4/b413500h/ for crystallographic data in .cif or other electronic format.
Acknowledgements
The help and assistance of Dr J. E. Davies of the Cambridge University Chemical Laboratory X-ray department is gratefully acknowledged. DSP thanks the Alfred Benzon Foundation and the Knud Højgaard Foundation for financial support.References
- W. A. Donaldson, Tetrahedron, 2001, 57, 8589 CrossRef CAS.
- H. Lebel, J.-F. Marcoux, C. Molinaro and A. B. Charette, Chem. Rev., 2003, 103, 977 CrossRef CAS.
- H. Nozaki, S. Moriuti, H. Takaya and R. Noyori, Tetrahedron Lett., 1966, 43, 5239 CrossRef.
- H. Nozaki, H. Takaya, S. Moriuti and R. Noyori, Tetrahedron, 1968, 24, 3655 CrossRef CAS.
- S. Vangveravong and D. E. Nichols, J. Org. Chem., 1995, 60, 3409 CrossRef CAS.
- D. A. Evans, K. A. Woerpel, M. M. Hinman and M. M. Faul, J. Am. Chem. Soc., 1991, 113, 726 CrossRef CAS.
- D. A. Evans, K. A. Woerpel and M. J. Scott, Angew. Chem., Int. Ed. Engl., 1992, 31, 430 CrossRef.
- S. Masamune and R. E. Lowenthal, Tetrahedron Lett., 1991, 50, 7373 CrossRef CAS.
- T. Miimi, T. Uchida, R. Irie and T. Katsuki, Tetrahedron Lett., 2000, 41, 3647 CrossRef CAS.
- M. P. Doyle, L. Zhou, A. B. Dyatkin and D. A. Rupper, Tetrahedron Lett., 1995, 36, 7579 CrossRef CAS.
- H. Takahashi, M. Yoshioka, M. Ohno and S. Kobayashi, Tetrahedron Lett., 1992, 33, 2575 CrossRef CAS.
- A. B. Charette and H. Juteau, Tetrahedron, 1997, 53, 16277 CrossRef CAS.
- T. Katsuki, H. Kitajima, Y. Aoki and K. Ito, Bull. Chem. Soc. Jpn., 1997, 70, 207 CAS.
- T. Katsuki, H. Kitajima, Y. Aoki and K. Ito, Chem. Lett., 1995, 1113 CAS.
- A. G. M. Barrett and K. J. Kasdorf, Chem. Commun., 1996, 325 RSC.
- L. Horner, H. Hoffmann and V. G. Toscano, Chem. Ber., 1962, 95, 536 CrossRef CAS.
- Imidazole also acts a general base catalyst in this type of reaction. P. M. Ayrey and S. Warren, Tetrahedron Lett., 1989, 30, 4581 Search PubMed.
- P. Wallace and S. Warren, J. Chem. Soc., Perkin Trans. 1, 1988, 2971 RSC.
- R. A. Izydore and R. G. Ghirardelli, J. Org. Chem., 1973, 38, 1790 CrossRef CAS.
- W. S. Wadsworth Jr. and W. D. Emmons, J. Am. Chem. Soc., 1961, 83, 1733 CrossRef CAS.
- E. E. Schweitzer and W. S. Creasy, J. Org. Chem., 1971, 36, 2379 CrossRef CAS.
- N. Feeder, G. Hutton, A. Nelson and S. Warren, J. Chem. Soc., Perkin Trans. 1, 1999, 3413 RSC.
- A. Nelson and S. Warren, Tetrahedron Lett., 1996, 37, 1501 CrossRef CAS.
- A. Nelson and S. Warren, J. Chem. Soc., Perkin Trans. I, 1999, 3425 RSC.
- T. Boesen, N. Feeder, M. E. Eastgate, D. J. Fox, J. A. Medlock, C. R. Tyzack and S. Warren, J. Chem. Soc., Perkin Trans. I, 2001, 118 RSC.
- J.-M. Poirier, L. Hennequin and M. Formani, Bull. Soc. Chim. Fr., 1986, 436 CAS.
- H. C. Kolb, M. S. VanNieuwenhze and K. B. Sharpless, Chem. Rev., 1994, 94, 2483 CrossRef CAS.
- L. Deng and E. N. Jacobsen, J. Org. Chem., 1992, 57, 4320 CrossRef CAS.
- D. J. Fox, D. S. Pedersen and S. Warren, Chem. Commun., 2004, 2598 RSC.
- R. S. Torr and S. Warren, J. Chem. Soc., Perkin Trans. I, 1983, 1173 RSC.
- D. S. Pedersen and C. Rosenbohm, Synthesis, 2001, 2431 CrossRef CAS.
- G. M. Sheldrick, SHELXS-97/SHELXL-97, University of Göttingen, Germany, 1997 Search PubMed.
| This journal is © The Royal Society of Chemistry 2005 |