
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSulfonated sucrose-derived carbon: efficient carbocatalysts for ester hydrolysis†
Guodong
Wen
 *,
Duo
Na
,
Yukun
Yan
and
Hongyang
Liu
*
*,
Duo
Na
,
Yukun
Yan
and
Hongyang
Liu
*
Shenyang National Laboratory for Materials Science, Institute of Metal Research, Chinese Academy of Sciences, Shenyang 110016, China. E-mail: wengd@imr.ac.cn; liuhy@imr.ac.cn
First published on 17th October 2024
Abstract
A series of sulfonated carbon acid catalysts with strong acidity was prepared by simultaneous carbonization and sulfonation of biomass sucrose in the presence of the organic sulfonating agent sulfosalicylic acid under hydrothermal conditions at temperatures ranging from 150 to 200 °C. It was found from FTIR and XPS spectra that the surface of carbon was efficiently functionalized with –SO3H groups. Research on the mechanism of the sulfonation process indicated that the intermediate 5-hydroxymethyl furfural (5-HMF), which was easily hydrolyzed from sucrose, was prone to carbonization and functionalized with –SO3H groups simultaneously. Compared with 5-HMF and fructose used as the initial carbon precursor, the slow hydrolysis of sucrose to intermediate 5-HMF to suppress its rapid carbonization is favorable for the efficient grafting of –SO3H groups when sucrose is used as the initial carbon precursor. The prepared sulfonated carbons were evaluated as acid catalysts in a typical ester hydrolysis reaction, namely, hydrolysis of ethyl acetate. The sulfonic acid groups were identified to be the active sites and quantified by a cation-exchange process. The activity of the sulfonated carbon was primarily correlated with the total number of active sites. However, when the total number of the –SO3H groups did not change, higher activities were shown on the sulfonated carbon with higher surface S content.
Introduction
Carbon-based structures (e.g., carbon dots, nanodiamonds, carbon spheres, mesoporous carbon, carbon nanotubes and graphene) have garnered wide attention because of their tremendous potential for various applications, such as laser materials, electrode materials and catalyst support.1–5 In the past several decades, it has been demonstrated that carbon could be used as important metal-free carbocatalysts in various kinds of heterogeneous reaction systems, including thermocatalysis (e.g., oxidation of alcohol,6 dehydrogenation of hydrocarbons,7 oxidation of cyclohexane,8 reduction of nitroarenes,9 amidation of esters,10 hydrochlorination of acetylene,11 and epoxidation of styrene12), photocatalysis and electrocatalysis. Due to the limited resource of metals, metal-free carbocatalysts are regarded as one of the most promising alternatives for designing efficient and sustainable catalytic systems to substitute metal catalysts.Currently, the research on solid acid catalysts is a hot topic because they (e.g., zeolites) are one of the most widely applied catalysts in many large-scale industrial processes such as hydroisomerization, catalytic reforming and catalytic cracking. To date, various novel solid acid catalysts have been developed. Metal chloride-functionalized Brønsted acidic ionic liquids containing Brønsted–Lewis acidic sites were rationally designed as efficient green acid catalysts to boost the conversion of biomass to fine chemicals.13,14 Carbon could also be used as a solid acid catalyst,15 and the surface physicochemical properties of the carbon could be well tuned by physical or chemical treatment. We have shown that the carboxylic acid groups could be enriched on the carbon via hydrothermal carbonization, and the obtained carbon could be efficiently used in acid-catalyzed reactions, such as Beckmann rearrangement reaction.16
The acidity of –SO3H groups is at par with H2SO4, which is by far one of the most important industrial chemicals. –SO3H groups could be grafted on carbon, and the obtained sulfonated carbons are regarded as a new class of metal-free carbocatalysts.17,18 Concentrated H2SO4 is involved in most of the processes developed for preparing sulfonated carbons, which are not safe, induce environmental hazards and have high energy requirements. In situ sulfonation routes with organic sulfonating agents instead of concentrated H2SO4 were developed to afford sulfonated carbon by the simultaneous sulfonation and hydrothermal carbonization of earth-abundant biomass saccharides, in which glucose was recognized as the preferred choice.17,19–21 This route offered obvious advantages in terms of environmental safety and energy consumption, and further study is needed to optimize the process.
In this work, sulfonated carbons were prepared via the in situ functionalization of –SO3H groups by the hydrothermal carbonization of biomass sucrose in the presence of sulfosalicylic acid at 150–200 °C. This process was systematically studied and optimized; the obtained carbons were tested in ester hydrolysis reaction, which is a kind of typical acid-catalyzed reaction. High activities were obtained over these sulfonated carbons.
Experimental
Materials
All chemicals were analytical grade and used without further purification. HY (SiO2/Al2O3 = 4.8) and HZSM-5 (SiO2/Al2O3 = 60) were provided from Dalian Institute of Chemical Physics, Chinese Academy of Sciences. The textural properties of HY and HZSM-5 are given in our previous report.16Catalysts preparation
In a typical process, 1.25 g sucrose, 5 g water and sulfosalicylic acid were mixed and sealed into a Teflon inlet in an autoclave and treated in an oven at 150 to 200 °C for 4 h. After the carbonization, the carbonaceous materials were washed with deionized water and ethanol (to remove physically adsorbed organic degradation products) and then dried at 80 °C overnight. The obtained sulfonated carbons carbonized at 150 to 200 °C for 4 h were denoted as SCx–y, where x is the carbonization temperature and y is the grams of the added sulfosalicylic acid.Catalysts characterization
The morphology and the particle size of the sulfonated carbons were visualized using scanning electron microscopy (SEM) with an FEI Quanta FEG 250. The energy-dispersive X-ray (EDX) mapping was also conducted on an FEI Quanta FEG 250. Transmission electron microscopy (TEM) images and STEM-EDX elemental maps were obtained by an FEI Tecnai G2 F20 microscope equipped with high-angle annular dark-field scanning TEM (HAADF-STEM) and energy dispersive X-ray detectors at an accelerating voltage of 200 kV. N2 adsorption–desorption was conducted on a Micrometrics 3Flex at 77 K to analyze the surface area, pore volume and pore size. The XRD patterns of the sulfonated carbon were given by a D/MAX-2500 PC X-ray diffractometer with monochromatized Cu Kα radiation (λ = 1.54 Å). A LabRam HR 800 equipment was used to record the Raman spectroscopy from 800 to 2000 cm−1 using a 633 nm laser. The X-ray photoelectron spectroscopy (XPS) measurements were performed on an ESCALAB 250 with an X-ray source of monochromatized Al Kα (1486.6 eV). The binding energies were corrected to the C 1s peak at 284.6 eV. FTIR experiments were conducted on a Cary 630 (Agilent Technologies) between 4000 and 400 cm−1 with a standard KBr disk method. Temperature-programmed desorption (TPD) was tested in helium with a flowrate of 50 mL min−1 at a heating rate of 10 °C min−1 from 80 to 900 °C, and the gas effluent was monitored online with mass spectrometry (OmniStar GSD 300). A cation exchange process was performed to quantify the S content in the sulfonated carbon. Specifically, 50 mg sulfonated carbon and 50 mg NaCl were added into 20 mL deionized water, and the mixture was stirred at room temperature for at least 24 h. Then, the pH value of the mixture was analyzed by a pH meter, and the S content (S) was calculated based on the following equation.| S = ([H+] − [H+]water) × V/m |
Catalytic tests
The hydrolysis of ethyl acetate was carried out in a thick-wall pressure vessel at 60 °C with magnetic stirring. In a typical experiment, 4 mL ethyl acetate, 60 g water, 600 μL 1,4-dioxane and 50 mg sulfonated carbon were added into the vessel. After the reaction, the liquid products were analyzed by GC (Agilent 8890) with an HP-5 column, and 1,4-dioxane added in the reaction mixture was used as the internal standard.Results and discussion
Catalyst characterization
The typical morphology of the sulfonated carbons is given in Fig. 1 (SEM images) and Fig. S1 (TEM images, ESI†). It could be seen from the SEM images that carbon spheres are uniformly formed via the hydrothermal carbonization, which is similar to our previous report.16 The average sphere size is 3.03 μm by counting 300 carbon spheres. The carbon sphere is generally obtained by the hydrothermal carbonization of biomass saccharides.1,16 Unlike the clean spheres that resulted directly from the carbonization of saccharides, the TEM images show that some small carbon particles are distributed on these sulfonated carbon spheres (Fig. S1, ESI†), which is likely to be caused by the addition of sulfosalicylic acid during the hydrothermal carbonization. As predicted, the carbons in the spheres exist in the amorphous form, as shown in the high-resolution TEM images, indicating that the degree of carbonization for the sulfonated carbon is low. The surface area of the sulfonated carbons is small (Table S1, ESI†), which is one of the typical features of hydrothermal carbon.17 | ||
| Fig. 1 SEM images of a typical sulfonated carbon (SC160-2.5). | ||
Only a wide peak ranging from 10 to 30° is shown in the XRD pattern of a typical sulfonated carbon (Fig. S2, ESI†), indicating that the sulfonated carbon is primarily composed of amorphous carbon. The Raman spectrum is given in Fig. S3 (ESI†) and is deconvoluted into four peaks, which is similar to that of Sadezky et al.22 and our previous works.15,16 The ID1/IG ratio, which is usually used to describe the defects density, is as high as 1.32, indicating that the degree of graphitization is low, and the carbon is rich in defects and functional groups. The Raman results are consistent with the TEM and XRD results.
The surface functional groups are detected by XPS analysis, and the results are given in Table 1 and Fig. S4 (ESI†). It could be found from Fig. S4 (ESI†) that the S 2p photoelectron peak is located at 168.5 eV, which is corresponding to the –SO3H groups,17,23,24 indicating that the samples are successfully functionalized with S species. The total number of the –SO3H groups quantified by a cation exchange process does not change significantly in the temperature range from 150 to 200 °C. However, obvious changes are shown on the surface S content via the XPS analysis. It could be seen from Table 1 that the surface S content increases with increasing carbonization temperature from 150 to 160 °C and then decreases with the further increase in the temperature ranging from 160 to 190 °C. The content of the surface oxygenated groups on the sulfonated carbons is very high, which is consistent with the typical characteristics of hydrothermal carbons caused by the low degree of carbonization in the carbon framework.1,16 According to our previous works,12,15,16 the O 1s spectrum is deconvoluted into three peaks at 531.4 eV, 532.5 eV and 533.7 eV, which are assigned to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O species, OC–O and OH species, respectively.
O species, OC–O and OH species, respectively.
| C (at%)a | O (at%)a | S (at%)a | –SO3H contentb (mmol g−1) | |
|---|---|---|---|---|
| a Measured by XPS. b Analyzed by cation exchange. | ||||
| SC150-0.625 | 79.32 | 20.22 | 0.46 | 0.16 |
| SC160-0.625 | 79.99 | 19.45 | 0.57 | 0.17 |
| SC170-0.625 | 79.67 | 19.86 | 0.47 | 0.16 |
| SC180-0.625 | 81.35 | 18.30 | 0.35 | 0.18 |
| SC190-0.625 | 81.70 | 18.06 | 0.25 | 0.19 |
| SC200-0.625 | 82.23 | 17.37 | 0.4 | 0.17 |
The grafting of –SO3H groups could also be confirmed by FTIR analysis (Fig. 2). The characteristic SO asymmetric (1211 and 1167 cm−1) and symmetric (1028 cm−1) stretching bands could be found in the figure.17 The signals for other functional groups are also shown in the spectrum. The wide peak at 3431 cm−1 corresponded to the stretching vibrations of OH groups.25 The peak at 2924 cm−1 is assigned to the stretching vibrations of sp3 C–H groups. A typical CO bending vibration is shown at 1704 cm−1, while a typical aromatic CC stretching vibration is found at 1621 cm−1.25 The peak at 1384 cm−1 is originated from the stretching vibration of OH in alcohol,26 and the peak at 802 cm−1 is caused by the C–H deformation of aromatic bonds.25 The FTIR results agree well with the XPS and Raman results. These results indicate that the sulfonated carbons are rich in various functional groups.
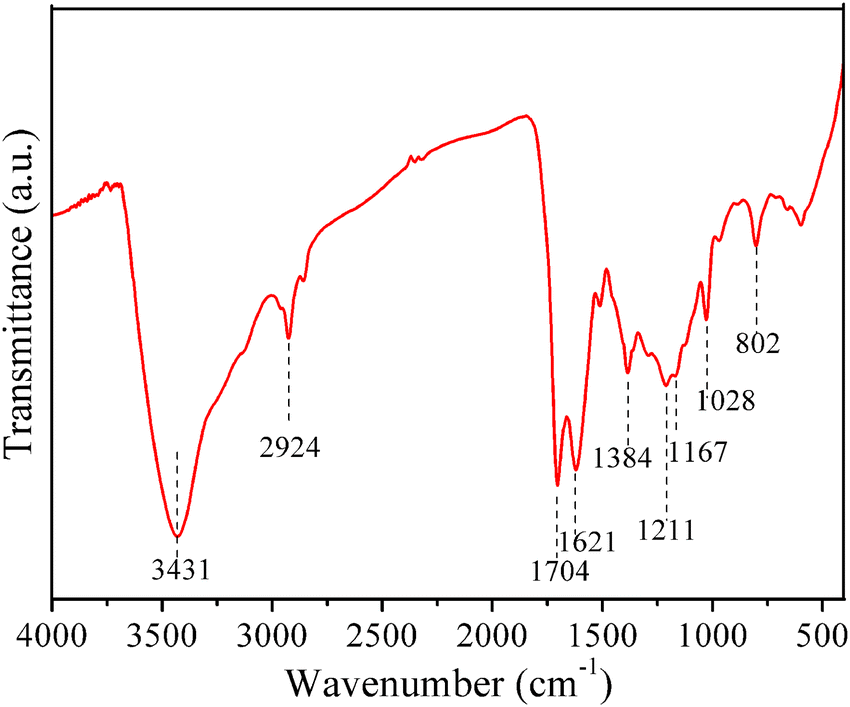 | ||
| Fig. 2 FTIR of a typical sulfonated carbon (SC160-2.5). | ||
TPD experiment was conducted to study the surface functional groups on sulfonated carbon (Fig. 3). The functionalization of –SO3H groups on the carbon could be deduced by the formation of SOx evolved during the TPD process.27 According to our previous work, the evolution profiles of CO2 could be deconvoluted into three peaks at about 260 °C (carboxylic acid), 300–500 °C (carboxylic anhydride), and 500–600 °C (lactone).15 The three oxygenated species could also be identified in the CO evolution profile based on their desorbed temperatures, which are similar to those in the CO2 profiles. Besides, one more peak at higher temperatures between 700 °C and 800 °C in the CO evolution profile was observed, which is originated from the phenol groups. With the increase in the temperatures from 80 to 900 °C, the carboxylic acid, carboxylic anhydride and lactone groups decompose to give both the CO2 and CO species, while only CO is obtained via the decomposition of phenol groups. These results are consistent with the XPS and FTIR results.
 | ||
| Fig. 3 Evolution profiles of the gas effluent with different components (a) and enlarged SOx signals (b) during the TPD for the typical sulfonated carbon (SC160-2.5). The CO2 (c) and CO (d) evolution profiles were deconvoluted into several peaks assigned to different oxygenated species. | ||
The distribution of the –SO3H groups on the carbon was analyzed by SEM-EDX mapping and high-resolution STEM-EDX mapping. It could be seen from Fig. S5 and S6 (ESI†) that the S species are homogeneously distributed on the sulfonated carbon. Only one S 2p photoelectron peak at 168.5 eV is given in Fig. S4 (ESI†), indicating that the S species existed in the form of –SO3H groups.17 No low-valent S species are found on the catalysts because of the absence of S 2p photoelectron peak at 164 eV corresponding to the –S–, –SH and C–S species. As a result, the –SO3H groups are homogeneously distributed on the surface of these micro-level carbon spheres.
Catalytic test for the hydrolysis of ethyl acetate
The hydrolysis of ethyl acetate is a typical acid-catalyzed ester hydrolysis reaction. The sulfonated carbons carbonized at different temperatures ranging from 150 to 200 °C, namely, SC-150-0.625, SC-160-0.625, SC-170-0.625, SC-180-0.625, SC-190-0.625 and SC-200-0.625, were selected as catalysts in the hydrolysis of ethyl acetate, and ethanol and ethyl acetic were the only detected products in the present research (Fig. S7, ESI†). It can be seen from Fig. 4 that the sulfonated carbon carbonized at 160 °C shows the maximum conversion. Carboxylic acid groups were generally considered as the primary acid sites on the pristine carbon,15 which could also be found on the sulfonated carbon via the XPS (Fig. S4, ESI†) and TPD (Fig. 3) analysis besides –SO3H groups. In our previous works, we showed that small organic molecules with specific functional groups could be efficiently used as model catalysts to mimic the catalytic roles of the functional groups on the carbon during the reactions.5,12,28 In order to identify the active sites on the sulfonated carbon for the ethyl acetate hydrolysis reaction, benzoic acid was selected as the catalyst to mimic carboxylic acid groups, while benzenesulfonic acid was chosen to mimic the –SO3H groups (Table S2, ESI†). It can be seen in Table S1 (ESI†) that the conversion on benzenesulfonic acid was significantly higher than that on benzoic acid, which was very similar to that observed in the blank experiment in the absence of any catalyst. These results from model catalysts indicated that –SO3H groups instead of carboxylic acid groups were the primary active sites for the hydrolysis of ethyl acetate. | ||
| Fig. 4 Influence of the carbonization temperature for preparing the sulfonated carbon on the catalytic performance of ethyl acetate hydrolysis. Reaction conditions: sulfonated carbon 50 mg, 60 mL water, 600 μL 1,4-dioxane, ethyl acetate 4 mL, 60 °C, 24 h. | ||
Although the total number of –SO3H sites does not change significantly in the temperature range from 150 to 200 °C, the highest surface S content is observed on SC-160-0.625. Therefore, the activity of the sulfonated carbon is primarily ascribed to the surface S sites when the total number of –SO3H sites does not change significantly.
The catalytic performance of the sulfonated carbon could be improved when more sulfosalicylic acid was added in the carbonization process. It can be seen from Fig. 5 that the conversion increases when the mass of sulfosalicylic acid is increased from 0.625 g to 2.5 g and then decreases with the further addition of sulfosalicylic acid. The surface S content on the SC-160-2.5 is 0.47 at%, which is a little lower than that on SC-160-0.625 (0.57 at%). However, the total number of –SO3H groups on SC-160-2.5 is much higher than that on SC-160-0.625 (Fig. S8, ESI†). These results indicate that the activity is primarily related to the total number of –SO3H sites when it changes obviously in the carbon.
 | ||
| Fig. 5 Influence of mass of sulfosalicyclic acid initially added in the carbonization process on the catalytic performance. Carbonization conditions: 1.25 g sucrose, 5 g H2O, 160 °C, 4 h. Reaction conditions: sulfonated carbon 50 mg, 60 mL water, 600 μL 1,4-dioxane, ethyl acetate 4 mL, 60 °C, 24 h. | ||
The hydrothermal carbonization process was carefully studied to give an insight into the carbonization mechanism. 109.8 mg sulfonated carbon was obtained when both 1.25 g sucrose and 0.625 g sulfosalicylic acid were added into 5 g water and then carbonized at 160 °C for 4 h. When only 1.25 g sucrose was added into 5 g water, 5.9 mg carbon product was obtained. Moreover, a clear solution was obtained after the carbonization when only 0.625 g sulfosalicylic acid was added into 5 g water. These results demonstrate that sulfosalicylic acid could not be carbonized at 160 °C but it significantly promoted the carbonization of sucrose. It is well known that sucrose could be easily hydrolyzed to glucose and fructose; thus, glucose and fructose were assumed as the intermediates during the carbonization, and they were added as the carbon precursors instead of sucrose. It was found that 222.1 mg sulfonated carbon was formed when fructose was used as the carbon precursor; however, only 13.1 mg carbon was obtained when glucose was used as the carbon precursor, indicating that fructose was prone to carbonization. Although glucose was recognized as the preferred choice in in situ sulfonation routes with organic sulfonating agents,17 the carbonization of glucose at a low temperature of 160 °C was severely suppressed. 5-Hydroxymethyl furfural (5-HMF) is generally regarded as the intermediate during the hydrothermal carbonization of biomass saccharides,29 and fructose tends to hydrolyze to 5-hydroxymethyl furfural as compared with glucose.
The sulfonated carbons from fructose and 5-HMF were tested in the hydrolysis reaction; however, the obtained conversions are lower than that on sucrose-derived carbon (Fig. 6), indicating that sucrose is suitable to be used as the carbon precursor. The total numbers of –SO3H sites on the sulfonated carbon derived from sucrose, fructose and 5-HMF were 0.17, 0.16 and 0.14 mmol g−1, respectively. It was assumed that the grafting of –SO3H groups is not efficient because the carbonization process is too fast when fructose or 5-HMF is initially added as the carbon precursor. Rapid carbonization led to a low S content in the sulfonated carbon; thus, lower conversions were observed on the carbons from fructose and 5-HMF.
 | ||
| Fig. 6 Influence of the initially added carbon precursors in the carbonization process on the catalytic performance. Reaction conditions: sulfonated carbon 50 mg, 60 mL water, 600 μL 1,4-dioxane, ethyl acetate 4 mL, 60 °C, 24 h. | ||
SC-160-2.5, which showed the best performance in the hydrolysis reaction, was tested as the typical sulfonated carbon for subsequent kinetic research. The conversion vs. reaction time diagram was obtained, which is given in Fig. 7. As shown in the figure, the conversion increases with the increase in the reaction time (Fig. 7a), and the reaction order for ethyl acetate concentration is pseudo first order (Fig. 7b).
 | ||
| Fig. 7 Influence of reaction time on the conversion over SC-160-2.5. Reaction conditions: 50 mg catalyst, 60 mL water, 600 μL 1,4-dioxane, ethyl acetate 4 mL, 60 °C. | ||
The activity of SC-160-2.5 was compared with the activities of other typical solid acid catalysts (e.g., HY and HZSM-5) (Fig. 8a). HY and HZSM-5 zeolites are used as acid catalysts in a wide range of acid-catalyzed reactions. It is shown that the conversion on SC-160-2.5 is similar to that on HZSM-5, which is much higher than that on HY.
 | ||
| Fig. 8 Hydrolysis of ethyl acetate over different solid acid catalysts. Reaction conditions: 50 mg catalyst, 60 mL water, 600 μL 1,4-dioxane, ethyl acetate 4 mL, 60 °C. The SC-160-2.5 (p) represents the sulfonated carbon prepared from p-toluenesulfonic acid. | ||
Other organic reagent containing –SO3H groups was also tested instead of sulfosalicylic acid during the hydrothermal carbonization to prepare the sulfonated carbon catalysts. p-Toluenesulfonic acid is widely used to prepare the sulfonated carbon;17 however, the activity of the carbon prepared from p-toluenesulfonic acid via our process is significantly lower than that of the carbon from sulfosalicylic acid (Fig. 8), indicating that sulfosalicylic acid is prone to be grafted on the carbon framework. It has been shown by our previous results that sulfosalicylic acid could significantly promote the carbonization of sucrose owing to the carboxylic acid and phenol groups on sulfosalicyclic acid, which are prone to interaction with the sucrose or intermediates as compared with the methyl groups on p-toluenesulfonic acid.
The catalyst was recycled to test the reusability of the sulfonated carbon. After the reaction, the catalyst was filtered, washed with water, and then dried at 80 °C overnight. Then, fresh reactants and internal standard were added to perform the next cycle of experiment. As shown in Fig. 9, the ethyl acetate conversion slowly decreases from 21.9% to 15.5% after the recycling, indicating that the sulfonated carbon prepared by the hydrothermal route is relatively stable and could be reused in the ester hydrolysis reaction system. The sulfonated carbon suffers from severe deactivation problem in many studies. Although the stability of our sulfonated carbon is not the best, it surpasses that of many reported sulfonated carbons.30–32 The number of –SO3H groups on the carbon after the recycling test quantified via the cation-exchange process was 15.5 mmol g−1, which is 70.8% of that in the fresh catalyst. Similar percentages left after the recycling test for the conversion and number of –SO3H groups indicated that the decrease in conversion was primarily related to the leaching of the –SO3H groups, which could be identified from the XPS (Fig. S9, ESI†) and FTIR spectra (Fig. S10, ESI†). The sulfonated carbon is still primarily composed of amorphous carbon after the recycling test as shown in the XRD pattern because of the presence of the wide peak ranging from 10 to 30° (Fig. S11, ESI†).
 | ||
| Fig. 9 Recycling test of SC-160-2.5 for the hydrolysis of ethyl acetate. Reaction conditions: 50 mg catalyst, 60 mL water, 600 μL 1,4-dioxane, ethyl acetate 4 mL, 60 °C, 24 h. | ||
Conclusions
Highly efficient sulfonated carbon acid catalysts were prepared by the simultaneous sulfonation and carbonization of biomass sucrose in the presence of organic sulfonating agent sulfosalicylic acid under hydrothermal conditions at different temperatures ranging from 150 to 200 °C. Concentrated H2SO4 is avoided as the sulfonating agent, which is not safe and induces environmental hazards. Sulfosalicylic acid was not only used as the organic sulfonating agent but also boosted the carbonization of sucrose. Research on the mechanism of the sulfonation process indicated that the intermediate 5-hydroxymethyl furfural (5-HMF), which was easily hydrolyzed from sucrose, was prone to carbonization and simultaneously functionalized with the –SO3H groups. Although glucose was recognized as the preferred choice in in situ sulfonation routes with organic sulfonating agents, the carbonization of glucose at a low temperature of 160 °C was severely suppressed. Compared with 5-HMF and fructose as the initial carbon precursors, the slow hydrolysis of sucrose to 5-HMF to suppress its rapid carbonization is favorable for the efficient grafting of –SO3H groups when sucrose is used as the initial carbon precursor. The activity of the sulfonated carbon was primarily related to the total number of active sites. However, when the total number of the –SO3H groups did not change significantly, higher activities were shown on the sulfonated carbon with higher surface S content. The activity of the sulfonated carbon was high, which is comparable with that of the conventional solid acid zeolite HZSM-5 and HY.Author contributions
Guodong Wen: funding acquisition, writing – original draft, validation, methodology, investigation, formal analysis, data curation. Duo Na: data curation, validation, writing – review and editing. Yukun Yan: validation, writing – review and editing. Hongyang Liu: validation, methodology, investigation.Data availability
The authors will supply the relevant data upon reasonable requests.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work is financially supported by National Key Research and Development Program of China (2023YFB3810600, 2022YFB4003100, 2022YFA1504500), National Natural Science Foundation of China (22072162, 92145301, U21B2092), International Partnership Program of Chinese Academy of Sciences (172GJHZ2022028MI), Natural Science Foundation of Liaoning Province (2021-MS-013) and Shenyang National Laboratory for Materials Science (L2019F30). This work is also supported by Guangxi Collaborative Innovation Centre of Structure and Property for New Energy and Materials, Science Research, Technology Development Project of Guilin (20210102-4).References
- S. J. Yu, J. K. He, Z. E. Zhang, Z. H. Sun, M. Y. Xie, Y. Q. Xu, X. Bie, Q. H. Li, Y. G. Zhang, M. Sevilla, M. M. Titirici and H. Zhou, Adv. Mater., 2024, 36, 2307412 CrossRef CAS PubMed.
- X. S. Zhang, T. Q. Cao, G. Y. Zhang, Q. Liu, G. Kong, K. J. Wang, Y. Jiang, X. Zhang and L. J. Han, J. Mater. Chem. A, 2024, 12, 4996–5039 RSC.
- Y. Q. Zhang and S. Y. Lu, Chem, 2024, 10, 134–171 CAS.
- H. X. Gong, S. C. Chen, J. B. H. Tok and Z. N. Bao, Matter, 2023, 6, 2206–2234 CrossRef CAS.
- D. S. Su, G. D. Wen, S. C. Wu, F. Peng and R. Schlögl, Angew. Chem., Int. Ed., 2017, 56, 936–964 CrossRef CAS PubMed.
- J. Meng, Z. H. Tong, H. X. Sun, Y. Z. Liu, S. Q. Zeng, J. N. Xu, Q. Q. Xia, Q. J. Pan, S. Dou and H. P. Yu, Adv. Sci., 2022, 9, 2200518 CrossRef CAS PubMed.
- M. Z. Yang, A. Lenarda, S. Frindy, Y. S. Sang, V. Oksanen, A. Bolognani, L. Hendrickx, J. Helaja and Y. D. Li, PNAS, 2023, 120, e2303564120 CrossRef CAS PubMed.
- H. Yu, F. Peng, J. Tan, X. W. Hu, H. J. Wang, J. Yang and W. X. Zheng, Angew. Chem., Int. Ed., 2011, 50, 3978–3982 CrossRef CAS PubMed.
- Y. J. Gao, D. Ma, C. L. Wang, J. Guan and X. H. Bao, Chem. Commun., 2011, 47, 2432–2434 RSC.
- K. P. Patel, E. M. Gayakwad and G. S. Shankarling, New J. Chem., 2020, 44, 2661–2668 RSC.
- B. Dai, K. Chen, Y. Wang, L. H. Kang and M. Y. Zhu, ACS Catal., 2015, 5, 2541–2547 CrossRef CAS.
- G. D. Wen, Q. Q. Gu, Y. F. Liu, R. Schlögl, C. X. Wang, Z. J. Tian and D. S. Su, Angew. Chem., Int. Ed., 2018, 57, 16898–16902 CrossRef CAS PubMed.
- Y. Y. Yu, Z. Q. Liu, G. Q. Zhang, Y. J. Liu and A. G. Ying, J. Cleaner Prod., 2022, 330, 129775 CrossRef CAS.
- Z. Q. Liu, J. L. Guo, R. H. Liang, F. X. Wang, Z. K. Li, Y. J. Liu and A. G. Ying, Chem. Eng. J., 2024, 479, 147757 CrossRef CAS.
- G. D. Wen, J. Y. Diao, S. C. Wu, W. M. Yang, R. Schlögl and D. S. Su, ACS Catal., 2015, 5, 3600–3608 CrossRef CAS.
- G. D. Wen, B. L. Wang, C. X. Wang, J. Wang, Z. J. Tian, R. Schlögl and D. S. Su, Angew. Chem., Int. Ed., 2017, 56, 600–604 CrossRef CAS PubMed.
- L. J. Konwar, P. Mäki-Arvela and J. P. Mikkola, Chem. Rev., 2019, 119, 11576–11630 CrossRef CAS PubMed.
- L. G. do Nascimento, I. M. Dias, G. B. M. de Souza, L. C. Mourão, M. B. Pereira, J. C. V. Viana, L. M. Lião, G. R. de Oliveira and C. G. Alonso, New J. Chem., 2022, 46, 6091–6102 RSC.
- I. F. Nata, C. Irawan, P. Mardina and C. K. Lee, J. Solid State Chem., 2015, 230, 163–168 CrossRef CAS.
- X. H. Qi, Y. F. Lian, L. L. Yan and R. L. Smith Jr, Catal. Commun., 2014, 57, 50–54 CrossRef CAS.
- J. F. Yang, H. Y. Zhang, Z. F. Ao and S. F. Zhang, Catal. Commun., 2019, 123, 109–113 CrossRef CAS.
- A. Sadezky, H. Muckenhuber, H. Grothe, R. Niessner and U. Pöschl, Carbon, 2005, 43, 1731–1742 CrossRef CAS.
- L. J. Konwar, A. Samikannu, P. Mäki-Arvela and J. P. Mikkola, Catal. Sci. Technol., 2018, 8, 2449–2459 RSC.
- K. Nakajima, M. Okamura, J. N. Kondo, K. Domen, T. Tatsumi, S. Hayashi and M. Hara, Chem. Mater., 2009, 21, 186–193 CrossRef CAS.
- Q. Pang, L. Q. Wang, H. Yang, L. S. Jia, X. W. Pan and C. C. Qiu, RSC Adv., 2014, 4, 41212–41218 RSC.
- M. M. Antunes, P. A. Russo, P. V. Wiper, J. M. Veiga, M. Pillinger, L. Mafra, D. V. Evtuguin, N. Pinna and A. A. Valente, ChemSusChem, 2014, 7, 804–812 CrossRef CAS PubMed.
- M. Hara, T. Yoshida, A. Takagaki, T. Takata, J. N. Kondo, S. Hayashi and K. Domen, Angew. Chem., Int. Ed., 2004, 43, 2955–2958 CrossRef CAS PubMed.
- S. C. Wu, G. D. Wen, X. M. Liu, B. W. Zhong and D. S. Su, ChemCatChem, 2014, 6, 1558–1561 CrossRef CAS.
- M. M. Titirici, R. J. White, C. Falco and M. Sevilla, Energy Environ. Sci., 2012, 5, 6796–6822 RSC.
- P. Q. Yan, Z. L. Xie, S. Y. Tian, F. Li, D. Wang, D. S. Su and W. Qi, RSC Adv., 2018, 8, 38150–38156 RSC.
- L. Tumkot, A. T. Quitain, P. Boonnoun, N. Laosiripojana, T. Kida and A. Shotipruk, ACS Omega, 2020, 5, 23542–23548 CrossRef CAS PubMed.
- J. M. Fraile, E. García-Bordejé, E. Pires and L. Roldán, J. Catal., 2015, 324, 107–118 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4nj03006k |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2024 |