Comparison of topological descriptors for similarity-based virtual screening using multiple bioactive reference structures†
Jérôme
Hert
a,
Peter
Willett
*a,
David J.
Wilton
a,
Pierre
Acklin
b,
Kamal
Azzaoui
b,
Edgar
Jacoby
b and
Ansgar
Schuffenhauer
b
aKrebs Institute for Biomolecular Research and Department of Information Studies, University of Sheffield, Western Bank, Sheffield, UK S10 2TN
bNovartis Institutes for BioMedical Research, Discovery Technologies, CH-4002, Basel, Switzerland
First published on 29th September 2004
Abstract
This paper reports a detailed comparison of a range of different types of 2D fingerprints when used for similarity-based virtual screening with multiple reference structures. Experiments with the MDL Drug Data Report database demonstrate the effectiveness of fingerprints that encode circular substructure descriptors generated using the Morgan algorithm. These fingerprints are notably more effective than fingerprints based on a fragment dictionary, on hashing and on topological pharmacophores. The combination of these fingerprints with data fusion based on similarity scores provides both an effective and an efficient approach to virtual screening in lead-discovery programmes.
Introduction
Virtual screening is widely used to enhance the cost-effectiveness of drug-discovery programmes, by ranking databases of chemical structures in decreasing probability of activity; this prioritisation then means that biological testing can be focused on just those few molecules that have significant a priori probabilities of activity.1, 2 There are many different ways in which a database can be prioritised; here, we focus on similarity searching methods.3, 4 Similarity searching is one of the most widely used virtual-screening approaches, and involves matching a known active molecule, the reference structure, against each of the database molecules, computing a measure of structural similarity in each case, and then ranking the database molecules in order of decreasing similarity score. Structurally similar molecules are likely to have similar biological activities5–7 and there is hence an extensive literature associated with the similarity measures that can be used to quantify the degree of resemblance between a reference structure and each of the database molecules.Most of the studies of similarity searching that have been reported thus far have considered the use of only a single bioactive reference structure. It is, however, increasingly the case that several, structurally diverse, reference structures may be available, e.g., published competitor compounds or hits from high-throughput screening (HTS), and this has stimulated interest in the use of multiple reference structures to identify further molecules for biological screening.8, 9 We have recently reported a detailed comparison of several different search algorithms that can be used in such circumstances, and identified two, data fusion and binary kernel discrimination (BKD), that provided a high level of effectiveness in simulated virtual screening experiments.10
An important component of any similarity procedure is the structure representation that is used to encode the molecules that are to be searched, with 2D fragment bit-strings (or fingerprints) of various types being by far the most commonly used in current chemoinformatics systems.11, 12 A fingerprint is a binary string that encodes the presence of substructural fragments, i.e. topological patterns of atoms and bonds, in a molecule. This is clearly a very simple representation of molecular structure but one that has been used with considerable success ever since the earliest reports of similarity searching,13, 14 and also for related chemoinformatics tasks such as molecular diversity analysis15 and database clustering.16 In two much-cited papers, Brown and Martin compared several different types of fingerprint when used for cluster-based physicochemical property prediction;17, 18 here, we report an analogous comparison of fingerprints when they are used for similarity-based virtual screening using multiple reference structures.
Results and discussion
Taking account of the different search algorithms, fingerprint types and normalisation schemes described in the experimental section, there is a total of 30 different similarity procedures available for evaluation. Each such procedure was used with each of 11 different activity classes from the MDL Drug Data Report (MDDR)19 database, with ten searches being carried out for the actives in each particular activity class. A different set of ten active reference structures was used for each of the ten searches, this set remaining constant across the 30 different similarity procedures. The results of the searches are shown in Tables 1–4, the first two listing the average recall obtained from the top 1% of the rankings for each of the activity classes, and the second two listing the average recall from the top 5% of the rankings. The mean recalls, averaged over all of the 11 activity classes, are shown in Figs. 1 and 2 (which also show the mean recall for data fusion and BKD averaged over all of the different fingerprint types considered here).| Activity classes | BCI | Daylight | Unity | Avalon | SimilogA | SimilogB | CATS | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Mean | SD | Mean | SD | Mean | SD | Mean | SD | Mean | SD | Mean | SD | Mean | SD | |
| 5HT3 antagonists | 35.5 | 3.3 | 27.6 | 5.0 | 29.0 | 6.1 | 34.9 | 4.8 | 34.6 | 11.3 | 41.3 | 10.6 | 11.8 | 2.5 |
| 5HT1A agonists | 27.4 | 6.4 | 21.7 | 4.9 | 19.5 | 3.9 | 21.3 | 5.7 | 16.2 | 5.4 | 20.1 | 6.2 | 9.0 | 2.1 |
| 5HT Reuptake inhibitors | 25.5 | 5.6 | 29.3 | 6.8 | 27.8 | 6.6 | 24.4 | 6.3 | 19.4 | 6.2 | 22.2 | 6.9 | 9.6 | 1.9 |
| D2 antagonists | 27.1 | 4.9 | 24.9 | 6.3 | 23.2 | 5.6 | 23.3 | 4.6 | 18.0 | 3.8 | 21.6 | 3.9 | 10.5 | 4.0 |
| Renin inhibitors | 65.1 | 8.7 | 61.6 | 7.3 | 62.3 | 5.5 | 63.0 | 4.4 | 32.4 | 18.9 | 32.3 | 15.7 | 43.0 | 10.4 |
| Angiotensin II AT1 antagonists | 47.1 | 2.3 | 47.7 | 2.6 | 47.2 | 2.1 | 45.9 | 2.6 | 36.8 | 6.7 | 36.1 | 4.6 | 38.6 | 6.4 |
| Thrombin inhibitors | 39.0 | 7.0 | 32.6 | 5.6 | 33.7 | 7.1 | 36.5 | 7.4 | 11.2 | 5.9 | 11.3 | 6.2 | 30.4 | 8.5 |
| Substance P antagonists | 32.0 | 6.1 | 33.0 | 5.5 | 31.7 | 4.0 | 22.6 | 4.4 | 15.7 | 6.5 | 15.4 | 6.9 | 8.0 | 3.2 |
| HIV protease inhibitors | 39.4 | 8.1 | 44.1 | 9.3 | 41.3 | 7.9 | 39.6 | 9.3 | 42.2 | 9.5 | 40.3 | 5.7 | 25.6 | 8.8 |
| Cyclooxygenase inhibitors | 24.6 | 5.1 | 21.2 | 5.3 | 20.8 | 4.2 | 21.0 | 2.8 | 16.2 | 2.7 | 16.5 | 3.6 | 10.5 | 2.5 |
| Protein kinase C inhibitors | 33.4 | 7.6 | 38.1 | 9.0 | 36.9 | 9.7 | 36.4 | 10.3 | 31.7 | 10.9 | 29.1 | 10.4 | 16.7 | 6.7 |
| Average over all classes | 36.0 | 5.9 | 34.7 | 6.1 | 34.0 | 5.7 | 33.5 | 5.7 | 24.9 | 8.0 | 26.0 | 7.3 | 19.4 | 5.2 |
| Activity classes | ECFP_2A | ECFP_2B | ECFP_4A | ECFP_4B | FCFP_2A | FCFP_2B | FCFP_4A | FCFP_4B | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Mean | SD | Mean | SD | Mean | SD | Mean | SD | Mean | SD | Mean | SD | Mean | SD | Mean | SD | |
| 5HT3 antagonists | 45.8 | 4.2 | 43.2 | 5.6 | 45.9 | 7.0 | 44.7 | 6.6 | 38.4 | 3.6 | 36.5 | 4.5 | 45.1 | 4.5 | 44.3 | 4.1 |
| 5HT1A agonists | 29.4 | 6.3 | 29.2 | 5.3 | 33.5 | 6.2 | 33.4 | 5.6 | 18.9 | 2.3 | 18.0 | 2.5 | 28.5 | 4.3 | 28.0 | 4.0 |
| 5HT Reuptake inhibitors | 31.8 | 5.9 | 31.2 | 4.9 | 32.8 | 6.1 | 31.0 | 6.4 | 29.6 | 4.0 | 28.9 | 4.4 | 33.6 | 6.9 | 33.5 | 7.1 |
| D2 antagonists | 28.6 | 6.2 | 28.7 | 5.3 | 30.4 | 6.7 | 30.5 | 6.6 | 20.7 | 4.8 | 20.7 | 4.5 | 29.1 | 6.9 | 28.4 | 6.6 |
| Renin inhibitors | 76.6 | 2.4 | 76.2 | 2.7 | 77.9 | 3.4 | 78.0 | 2.3 | 49.5 | 6.5 | 48.9 | 6.8 | 71.1 | 7.6 | 71.2 | 8.0 |
| Angiotensin II AT1 antagonists | 49.0 | 3.0 | 50.0 | 3.0 | 49.6 | 3.4 | 49.8 | 3.0 | 44.9 | 2.2 | 45.2 | 1.9 | 48.3 | 3.2 | 48.7 | 3.2 |
| Thrombin inhibitors | 43.9 | 11.9 | 43.5 | 12.0 | 42.0 | 9.4 | 41.4 | 9.3 | 34.9 | 8.1 | 34.1 | 7.5 | 39.9 | 6.7 | 39.7 | 6.3 |
| Substance P antagonists | 35.6 | 6.3 | 37.0 | 6.0 | 38.3 | 9.3 | 37.4 | 9.2 | 28.3 | 5.3 | 27.7 | 5.5 | 35.3 | 6.4 | 34.7 | 6.3 |
| HIV protease inhibitors | 52.4 | 8.5 | 51.6 | 7.6 | 55.3 | 8.6 | 56.3 | 7.9 | 34.9 | 7.0 | 35.3 | 8.0 | 44.8 | 8.3 | 44.6 | 8.5 |
| Cyclooxygenase inhibitors | 23.1 | 3.3 | 22.1 | 3.3 | 22.8 | 3.6 | 22.4 | 3.7 | 20.5 | 3.3 | 20.3 | 3.2 | 24.5 | 5.0 | 23.8 | 4.6 |
| Protein kinase C inhibitors | 37.5 | 10.9 | 37.7 | 10.0 | 40.0 | 11.9 | 40.1 | 9.8 | 35.9 | 7.4 | 34.9 | 9.7 | 40.3 | 11.5 | 40.1 | 11.3 |
| Average over all classes | 41.2 | 6.3 | 40.9 | 6.0 | 42.6 | 6.9 | 42.3 | 6.4 | 32.4 | 5.0 | 31.9 | 5.3 | 40.0 | 6.5 | 39.7 | 6.4 |
| Activity classes | BCI | Daylight | Unity | Avalon | SimilogA | SimilogB | CATS | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Mean | SD | Mean | SD | Mean | SD | Mean | SD | Mean | SD | Mean | SD | Mean | SD | |
| 5HT3 antagonists | 35.5 | 4.1 | 31.9 | 2.8 | 31.0 | 5.3 | 30.5 | 4.5 | 34.4 | 3.6 | 34.7 | 4.6 | 8.7 | 3.2 |
| 5HT1A agonists | 31.0 | 5.5 | 22.6 | 3.9 | 20.1 | 4.3 | 19.4 | 5.3 | 26.2 | 4.2 | 27.3 | 4.6 | 10.1 | 2.6 |
| 5HT Reuptake inhibitors | 28.7 | 4.4 | 30.9 | 5.5 | 29.0 | 4.8 | 31.8 | 3.4 | 27.9 | 5.9 | 28.0 | 5.5 | 9.4 | 2.9 |
| D2 antagonists | 27.9 | 3.7 | 25.1 | 4.9 | 22.7 | 4.5 | 23.4 | 4.8 | 30.2 | 4.1 | 29.0 | 4.6 | 10.4 | 3.8 |
| Renin inhibitors | 53.0 | 8.3 | 51.3 | 3.3 | 50.2 | 3.2 | 56.7 | 4.0 | 43.6 | 15.4 | 48.6 | 13.8 | 60.6 | 2.0 |
| Angiotensin II AT1 antagonists | 43.1 | 3.0 | 43.4 | 3.0 | 41.4 | 4.0 | 43.0 | 4.7 | 44.2 | 1.8 | 45.0 | 1.8 | 40.2 | 2.2 |
| Thrombin inhibitors | 36.3 | 9.3 | 27.6 | 7.4 | 27.7 | 9.2 | 34.2 | 9.9 | 17.7 | 9.8 | 20.4 | 9.9 | 22.5 | 14.0 |
| Substance P antagonists | 23.8 | 5.8 | 24.7 | 5.1 | 23.1 | 4.6 | 18.0 | 4.8 | 19.7 | 6.8 | 20.2 | 6.6 | 9.3 | 2.3 |
| HIV protease inhibitors | 25.6 | 6.8 | 33.1 | 8.2 | 33.8 | 7.2 | 33.1 | 7.9 | 39.5 | 5.2 | 39.8 | 4.7 | 27.9 | 7.4 |
| Cyclooxygenase inhibitors | 22.6 | 6.4 | 19.8 | 4.9 | 18.5 | 4.8 | 19.0 | 4.7 | 17.2 | 1.3 | 16.6 | 1.6 | 7.8 | 1.2 |
| Protein kinase C inhibitors | 33.9 | 8.9 | 41.3 | 8.2 | 39.2 | 6.6 | 38.4 | 6.3 | 27.7 | 9.4 | 27.8 | 9.3 | 16.0 | 4.8 |
| Average over all classes | 32.8 | 6.0 | 32.0 | 5.2 | 30.6 | 5.3 | 31.6 | 5.5 | 29.8 | 6.2 | 30.7 | 6.1 | 20.3 | 4.2 |
| Activity classes | ECFP_2A | ECFP_2B | ECFP_4A | ECFP_4B | FCFP_2A | FCFP_2B | FCFP_4A | FCFP_4B | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Mean | SD | Mean | SD | Mean | SD | Mean | SD | Mean | SD | Mean | SD | Mean | SD | Mean | SD | |
| 5HT3 antagonists | 47.3 | 3.4 | 46.0 | 3.1 | 52.2 | 5.0 | 50.8 | 5.0 | 32.8 | 2.8 | 30.7 | 3.0 | 44.2 | 3.8 | 44.0 | 3.5 |
| 5HT1A agonists | 30.9 | 5.6 | 30.8 | 5.3 | 35.8 | 4.4 | 35.0 | 3.9 | 20.2 | 1.4 | 20.1 | 1.9 | 29.2 | 3.3 | 29.2 | 3.1 |
| 5HT Reuptake inhibitors | 29.9 | 4.4 | 30.1 | 4.4 | 31.9 | 5.8 | 32.0 | 5.7 | 26.4 | 6.6 | 25.2 | 7.2 | 32.7 | 6.2 | 33.1 | 6.1 |
| D2 antagonists | 28.0 | 6.2 | 27.6 | 6.2 | 31.8 | 5.4 | 31.9 | 5.8 | 21.1 | 4.4 | 20.4 | 5.0 | 29.7 | 5.5 | 29.2 | 5.2 |
| Renin inhibitors | 72.2 | 4.8 | 72.1 | 3.8 | 75.1 | 4.6 | 75.0 | 4.0 | 45.8 | 5.3 | 44.9 | 5.6 | 65.6 | 6.5 | 65.6 | 6.9 |
| Angiotensin II AT1 antagonists | 47.8 | 3.8 | 47.7 | 4.4 | 50.0 | 4.0 | 51.1 | 3.7 | 41.0 | 3.5 | 41.9 | 3.7 | 49.3 | 3.3 | 49.5 | 3.3 |
| Thrombin inhibitors | 41.9 | 11.7 | 41.3 | 11.1 | 42.0 | 10.6 | 42.1 | 10.0 | 27.3 | 7.8 | 26.6 | 7.8 | 37.9 | 9.4 | 37.1 | 8.4 |
| Substance P antagonists | 32.8 | 6.3 | 32.8 | 6.4 | 36.5 | 7.7 | 36.1 | 8.3 | 20.0 | 4.0 | 19.7 | 3.6 | 30.4 | 6.6 | 30.1 | 6.4 |
| HIV protease inhibitors | 48.7 | 6.8 | 49.2 | 6.6 | 54.3 | 7.5 | 54.1 | 6.7 | 28.6 | 6.6 | 28.9 | 7.2 | 43.4 | 8.1 | 42.9 | 8.0 |
| Cyclooxygenase inhibitors | 21.6 | 3.9 | 21.4 | 3.9 | 24.3 | 3.9 | 23.7 | 4.5 | 17.6 | 3.3 | 17.2 | 3.3 | 24.0 | 4.5 | 23.3 | 4.4 |
| Protein kinase C inhibitors | 41.0 | 7.4 | 41.9 | 6.9 | 46.7 | 8.9 | 46.3 | 8.1 | 33.2 | 9.5 | 32.6 | 9.5 | 45.3 | 9.1 | 44.9 | 8.9 |
| Average over all classes | 40.2 | 5.8 | 40.1 | 5.6 | 43.7 | 6.2 | 43.5 | 6.0 | 28.5 | 5.0 | 28.0 | 5.3 | 39.2 | 6.0 | 39.0 | 5.8 |
| Activity classes | BCI | Daylight | Unity | Avalon | SimilogA | SimilogB | CATS | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Mean | SD | Mean | SD | Mean | SD | Mean | SD | Mean | SD | Mean | SD | Mean | SD | |
| 5HT3 antagonists | 55.7 | 4.4 | 46.3 | 6.7 | 52.1 | 8.4 | 52.4 | 6.1 | 59.5 | 13.7 | 65.1 | 12.9 | 28.8 | 2.5 |
| 5HT1A agonists | 50.0 | 10.3 | 43.4 | 7.6 | 38.2 | 7.1 | 40.2 | 7.3 | 45.7 | 10.7 | 52.0 | 9.8 | 22.9 | 5.7 |
| 5HT Reuptake inhibitors | 42.6 | 8.9 | 44.6 | 10.5 | 45.4 | 8.7 | 40.6 | 11.0 | 38.3 | 9.1 | 45.0 | 8.3 | 25.7 | 5.2 |
| D2 antagonists | 46.5 | 8.5 | 42.4 | 9.7 | 38.6 | 7.5 | 35.9 | 6.0 | 45.7 | 7.5 | 52.0 | 5.0 | 21.7 | 6.5 |
| Renin inhibitors | 93.2 | 3.7 | 92.5 | 2.8 | 93.3 | 1.4 | 94.0 | 2.9 | 81.3 | 8.7 | 83.0 | 5.7 | 89.7 | 2.7 |
| Angiotensin II AT1 antagonists | 90.9 | 2.3 | 88.9 | 3.4 | 84.5 | 6.6 | 80.6 | 4.2 | 71.5 | 9.9 | 71.0 | 8.1 | 75.7 | 5.1 |
| Thrombin inhibitors | 69.0 | 5.3 | 61.3 | 8.1 | 63.0 | 7.6 | 69.2 | 7.3 | 37.4 | 13.0 | 37.1 | 11.5 | 60.8 | 8.4 |
| Substance P antagonists | 51.9 | 9.1 | 57.0 | 5.2 | 58.4 | 8.3 | 44.5 | 8.0 | 36.1 | 8.7 | 33.1 | 8.6 | 19.4 | 4.6 |
| HIV protease inhibitors | 66.5 | 6.7 | 67.3 | 9.8 | 68.4 | 8.3 | 60.9 | 12.8 | 70.4 | 8.3 | 69.9 | 6.8 | 60.9 | 9.2 |
| Cyclooxygenase inhibitors | 36.0 | 6.6 | 32.3 | 5.5 | 33.1 | 4.6 | 32.0 | 4.2 | 30.8 | 6.4 | 30.9 | 6.8 | 22.5 | 2.9 |
| Protein kinase C inhibitors | 45.2 | 7.9 | 48.4 | 9.5 | 49.2 | 11.0 | 47.9 | 8.1 | 45.7 | 9.6 | 46.0 | 8.3 | 30.0 | 7.3 |
| Average over all classes | 58.9 | 6.7 | 56.8 | 7.2 | 56.7 | 7.2 | 54.4 | 7.1 | 51.1 | 9.6 | 53.2 | 8.3 | 41.7 | 5.5 |
| Activity classes | ECFP_2A | ECFP_2B | ECFP_4A | ECFP_4B | FCFP_2A | FCFP_2B | FCFP_4A | FCFP_4B | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Mean | SD | Mean | SD | Mean | SD | Mean | SD | Mean | SD | Mean | SD | Mean | SD | Mean | SD | |
| 5HT3 antagonists | 67.6 | 6.2 | 63.1 | 8.5 | 65.3 | 6.7 | 63.4 | 7.6 | 62.7 | 6.9 | 61.6 | 7.4 | 68.6 | 6.7 | 67.7 | 6.6 |
| 5HT1A agonists | 54.4 | 8.1 | 52.8 | 6.7 | 58.7 | 7.9 | 57.5 | 7.7 | 45.1 | 5.3 | 44.8 | 4.1 | 55.1 | 7.2 | 54.1 | 6.1 |
| 5HT Reuptake inhibitors | 50.5 | 4.0 | 49.1 | 2.9 | 50.3 | 5.9 | 49.0 | 6.5 | 49.8 | 3.7 | 50.4 | 4.0 | 50.5 | 5.7 | 50.1 | 7.0 |
| D2 antagonists | 50.4 | 8.5 | 51.5 | 7.3 | 55.2 | 9.5 | 54.2 | 9.0 | 46.2 | 9.5 | 45.1 | 8.7 | 52.6 | 8.5 | 51.7 | 9.2 |
| Renin inhibitors | 97.6 | 0.6 | 97.6 | 0.5 | 96.7 | 0.8 | 97.0 | 0.8 | 92.1 | 4.8 | 92.9 | 3.9 | 97.6 | 1.3 | 97.6 | 1.1 |
| Angiotensin II AT1 antagonists | 97.0 | 1.7 | 96.9 | 1.7 | 98.0 | 0.8 | 97.8 | 0.6 | 86.8 | 5.5 | 87.3 | 5.2 | 95.0 | 4.7 | 95.1 | 4.1 |
| Thrombin inhibitors | 77.6 | 8.7 | 75.1 | 10.0 | 74.8 | 8.8 | 74.1 | 8.0 | 69.9 | 5.9 | 67.9 | 7.0 | 74.1 | 6.9 | 73.8 | 6.4 |
| Substance P antagonists | 62.0 | 8.2 | 62.7 | 7.7 | 67.3 | 9.5 | 65.5 | 10.2 | 48.8 | 6.5 | 48.1 | 8.1 | 59.8 | 7.9 | 59.1 | 9.1 |
| HIV protease inhibitors | 79.3 | 7.7 | 79.1 | 7.1 | 80.8 | 6.0 | 80.8 | 5.0 | 62.7 | 9.5 | 63.4 | 10.0 | 70.5 | 11.8 | 70.6 | 12.2 |
| Cyclooxygenase inhibitors | 36.0 | 5.4 | 34.6 | 5.3 | 34.4 | 5.4 | 32.7 | 6.2 | 36.7 | 4.7 | 35.6 | 4.8 | 36.9 | 6.2 | 36.5 | 6.8 |
| Protein kinase C inhibitors | 49.2 | 11.4 | 49.9 | 11.2 | 49.6 | 13.7 | 50.3 | 8.9 | 48.2 | 7.8 | 49.4 | 8.1 | 49.7 | 12.7 | 49.1 | 12.1 |
| Average over all classes | 65.6 | 6.4 | 64.8 | 6.3 | 66.5 | 6.8 | 65.7 | 6.4 | 59.0 | 6.4 | 58.8 | 6.5 | 64.6 | 7.2 | 64.1 | 7.3 |
| Activity classes | BCI | Daylight | Unity | Avalon | SimilogA | SimilogB | CATS | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Mean | SD | Mean | SD | Mean | SD | Mean | SD | Mean | SD | Mean | SD | Mean | SD | |
| 5HT3 antagonists | 58.8 | 5.9 | 51.3 | 3.5 | 49.0 | 5.4 | 52.4 | 4.1 | 49.5 | 7.7 | 48.8 | 8.4 | 20.9 | 6.2 |
| 5HT1A agonists | 54.7 | 5.8 | 40.9 | 5.6 | 37.2 | 4.1 | 34.7 | 4.7 | 48.8 | 6.1 | 49.8 | 7.1 | 23.0 | 3.6 |
| 5HT Reuptake inhibitors | 45.4 | 4.7 | 46.9 | 5.4 | 49.7 | 5.5 | 47.6 | 5.2 | 46.6 | 6.1 | 46.7 | 6.0 | 23.2 | 6.4 |
| D2 antagonists | 48.3 | 4.4 | 42.4 | 6.6 | 37.4 | 4.9 | 33.3 | 6.0 | 53.1 | 7.7 | 50.3 | 7.2 | 21.8 | 5.1 |
| Renin inhibitors | 93.5 | 1.3 | 90.1 | 2.0 | 88.6 | 1.9 | 90.0 | 4.0 | 85.5 | 2.7 | 87.5 | 2.6 | 91.1 | 1.5 |
| Angiotensin II AT1 antagonists | 86.3 | 3.5 | 86.9 | 2.0 | 80.5 | 6.1 | 82.0 | 4.6 | 82.0 | 4.8 | 84.1 | 4.6 | 71.2 | 4.8 |
| Thrombin inhibitors | 66.6 | 5.6 | 56.5 | 7.6 | 58.6 | 9.0 | 63.3 | 8.6 | 35.7 | 10.9 | 39.7 | 10.9 | 43.3 | 16.0 |
| Substance P antagonists | 44.8 | 7.2 | 51.8 | 6.3 | 47.1 | 5.2 | 39.9 | 3.7 | 36.8 | 7.6 | 36.5 | 7.0 | 24.1 | 2.7 |
| HIV protease inhibitors | 59.0 | 4.6 | 58.7 | 7.0 | 61.6 | 7.9 | 56.1 | 7.4 | 63.5 | 4.5 | 63.5 | 4.6 | 56.7 | 10.1 |
| Cyclooxygenase inhibitors | 33.4 | 7.8 | 29.9 | 7.8 | 26.5 | 7.2 | 30.9 | 6.7 | 28.1 | 4.4 | 28.0 | 4.2 | 15.9 | 1.5 |
| Protein kinase C inhibitors | 47.3 | 9.4 | 48.9 | 8.3 | 48.0 | 9.0 | 52.3 | 5.2 | 37.8 | 8.6 | 38.0 | 8.8 | 28.0 | 5.2 |
| Average over all classes | 58.0 | 5.5 | 54.9 | 5.6 | 53.1 | 6.0 | 53.0 | 5.5 | 51.6 | 6.5 | 52.1 | 6.5 | 38.1 | 5.8 |
| Activity classes | ECFP_2A | ECFP_2B | ECFP_4A | ECFP_4B | FCFP_2A | FCFP_2B | FCFP_4A | FCFP_4B | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Mean | SD | Mean | SD | Mean | SD | Mean | SD | Mean | SD | Mean | SD | Mean | SD | Mean | SD | |
| 5HT3 antagonists | 69.7 | 4.8 | 68.5 | 4.9 | 72.2 | 6.1 | 70.4 | 6.4 | 54.5 | 3.3 | 53.2 | 3.2 | 66.5 | 5.7 | 65.8 | 5.2 |
| 5HT1A agonists | 55.7 | 6.5 | 55.7 | 6.3 | 64.2 | 4.5 | 63.2 | 4.6 | 45.5 | 3.9 | 45.8 | 4.0 | 55.4 | 4.5 | 55.7 | 3.9 |
| 5HT Reuptake inhibitors | 48.1 | 4.0 | 47.9 | 2.8 | 49.7 | 5.7 | 49.5 | 4.2 | 46.0 | 5.7 | 44.9 | 5.3 | 51.1 | 5.2 | 50.3 | 5.6 |
| D2 antagonists | 50.3 | 6.0 | 48.9 | 5.3 | 56.1 | 5.4 | 54.7 | 5.9 | 44.4 | 5.9 | 45.6 | 6.1 | 50.6 | 6.2 | 51.2 | 5.8 |
| Renin inhibitors | 97.3 | 0.7 | 97.4 | 0.6 | 96.8 | 0.7 | 96.9 | 0.9 | 88.6 | 3.1 | 87.6 | 3.5 | 97.2 | 0.9 | 97.0 | 0.7 |
| Angiotensin II AT1 antagonists | 95.6 | 2.1 | 95.1 | 2.3 | 97.4 | 0.7 | 97.1 | 0.7 | 83.7 | 4.1 | 83.8 | 3.8 | 94.9 | 2.5 | 95.0 | 2.6 |
| Thrombin inhibitors | 74.0 | 6.6 | 71.7 | 6.8 | 74.7 | 6.4 | 72.5 | 7.3 | 58.9 | 6.9 | 58.1 | 6.7 | 70.2 | 4.2 | 70.1 | 4.3 |
| Substance P antagonists | 55.2 | 7.2 | 54.4 | 8.1 | 62.2 | 7.3 | 61.4 | 7.6 | 37.3 | 6.4 | 36.9 | 6.0 | 53.5 | 7.0 | 53.0 | 6.7 |
| HIV protease inhibitors | 76.2 | 4.5 | 76.9 | 4.6 | 80.0 | 4.7 | 79.4 | 4.8 | 59.0 | 6.8 | 58.4 | 6.5 | 71.9 | 8.9 | 70.7 | 8.3 |
| Cyclooxygenase inhibitors | 34.8 | 6.1 | 34.1 | 6.1 | 40.1 | 6.2 | 38.8 | 5.7 | 29.8 | 5.4 | 30.0 | 5.4 | 37.8 | 6.7 | 37.0 | 5.8 |
| Protein kinase C inhibitors | 52.9 | 7.1 | 53.9 | 7.0 | 57.8 | 6.7 | 57.0 | 6.6 | 45.4 | 10.0 | 45.5 | 9.9 | 54.6 | 7.4 | 55.2 | 7.6 |
| Average over all classes | 64.5 | 5.1 | 64.1 | 5.0 | 68.3 | 4.9 | 67.4 | 5.0 | 53.9 | 5.6 | 53.6 | 5.5 | 64.0 | 5.4 | 63.7 | 5.1 |
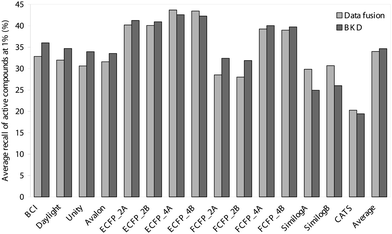 | ||
| Fig. 1 Comparison of the average recalls obtained in the top 1% of the ranked test-set using BKD and data fusion. | ||
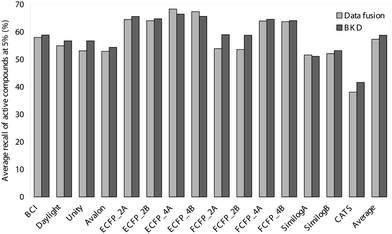 | ||
| Fig. 2 Comparison of the average recalls obtained in the top 5% of the ranked test-set using BKD and data fusion. | ||
Inspection of these tables reveals the very marked superiority of the circular substructure descriptors; indeed, there was only a single case where one of these fingerprints did not provide the best result, viz. the average recall at 1% using BKD for the set of cyclooxygenase inhibitors. This general effectiveness of the circular substructures (with the notable exception of FCFP_2) is highlighted in Figs. 1 and 2. Of these circular substructure fingerprints, the ECFP_4 ones, irrespective of the normalisation method (method A or method B) or of the search algorithm (data fusion or BKD), are the descriptors of choice for virtual screening of the sort advocated here. The FCFP_4 and ECFP_2 descriptors are also very effective: the former fingerprints seem to perform relatively better with the more heterogeneous (i.e., low self-similarity) classes, such as the cyclooxygenase and protein kinase C inhibitors, while the ECFP_2 fingerprints yield better results with the more homogeneous (i.e., high self-similarity) classes, such as the renin inhibitors.
As an alternative way of considering the figures in Tables 1–4, consider the enrichment factors20 to which these results correspond. The enrichment factor is the number of times better (in terms of active molecules retrieved) that a particular search algorithm is than a random selection of molecules from the database. Thus, the average enrichment values for ECFP_4B at 1% are 42.3 and 43.5 for BKD and data fusion, respectively, with the corresponding 5% values being 13.1 and 13.5, respectively, demonstrating the utility of the methods discussed here for virtual screening purposes.
Circular substructures of various sorts have been widely used for applications such as structure and substructure searching,21–23 constitutional symmetry.24 structure elucidation25 and, most recently, probabilistic modelling of bioactivity where a full training-set is available.26, 27 The work reported here demonstrates that this type of fragment is also very well suited to virtual screening using multiple reference structures.
When comparing the normalisation methods used for the circular substructure representations (see experimental section), method A, where all the initial features are just assigned a new bit-position, always provides descriptors that are more effective than method B. However, the differences are generally very small, and we would hence recommend the use of method B for the processing of these descriptors as this method is faster and, more importantly, is reproducible over different databases. There is little to choose between data fusion and BKD over the entire class of circular substructures, although it does appear that the use of these substructures with data fusion was particularly successful for the more heterogeneous classes like the cyclooxygenase and protein kinase C inhibitors. Conversely, when these descriptors were used with BKD, they worked particularly well for the more homogeneous activity classes, such as the renin inhibitors and the angiotensin II AT1 antagonists.
The dictionary-based descriptors, represented here by the BCI fingerprints, were ranked second overall, returning generally higher recalls than the hashed fingerprints, i.e., Unity, Daylight and Avalon. This finding is in agreement with the studies of cluster-based property prediction by Brown and Martin17, 18 (although they used different types of dictionary and hashed fingerprints from those studied here).
Perhaps our most surprising finding is the performance of the pharmacophore descriptors, with both the CATS and Similog fingerprints yielding consistently poorer recall values. Previous studies of these descriptors, for chemogenomics and scaffold-hopping applications,9, 28, 29 have demonstrated that they can be highly effective in operation, but this was certainly not the case for the present application. We note in the experimental section that both of these molecular characterisations are based on the encoding of the occurrences, rather than the incidences, of substructural fragments in a molecule, yielding an integer vector rather than a binary fingerprint. Here, however, they have been encoded in a binary form since the kernel function used in our BKD implementation requires a binary string. It is hence possible that the poor performance of the two pharmacophore fingerprints arose from the use of an inappropriate encoding mechanism. To test this, searches were carried out with the original, occurrence-based vectors; these searches used just data fusion, as this search algorithm does not necessarily require binary fingerprints for the generation of the ranked sets of scores that are fused together. Specifically, the rankings for the individual reference structures were computed using the non-binary form of the Tanimoto similarity coefficient and the Floersheim distance, as defined in the experimental section below. The use of the integer vectors and the non-binary coefficients did not improve the recall of the data fusion searches, and we hence conclude that the use of binary representations does not explain the poor performance of these 2D pharmacophore descriptors that is observed in Tables 1–4. It is perhaps worth noting in passing that previous comparisons of 2D fingerprints with 3D pharmacophore descriptors have often shown the former to be superior,17, 18, 30 despite the claimed effectiveness of the latter methods for diversity analysis and similarity searching.31
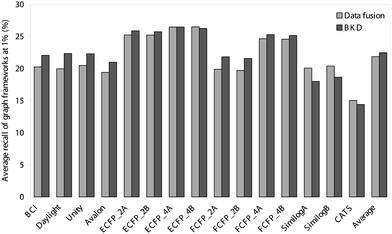
Thus far, we have evaluated the various approaches solely in terms of the numbers of active molecules that have been retrieved. It is, however, also of importance to consider the diversity of these sets of retrieved actives, since it is clearly preferable for the outputs also to maximise the numbers of chemotypes that are identified. We have hence analysed the outputs summarised in Tables 1–4 and Figs. 1 and 2 in terms of the numbers of distinct ring systems identified in the sets of retrieved actives. We have considered two levels of ring description, as illustrated in Fig. 3, and as discussed previously by Bemis and Murcko32 and by Xu and Johnson;33, 34 these authors refer to these levels of description as atomic frameworks or cyclic systems (Fig. 3a), and molecular frameworks or skeletal cyclic systems (Fig. 3b), respectively. Fig. 4 shows the percentages of the atomic frameworks in the complete set of actives that are retrieved in the top 1% of the ranking by each of the search procedures when averaged over all of the activity classes (i.e., as in Fig. 1); Fig. 5 gives the top 1% distribution for the molecular frameworks and Figs. 6 and 7 the corresponding top 5% distributions. It will be seen that the relative performance of the various procedures in terms of retrieving chemotypes (and hence in their suitability for scaffold-hopping applications) mirrors closely the relative performance based on numbers of actives (as shown in Figs. 1 and 2).
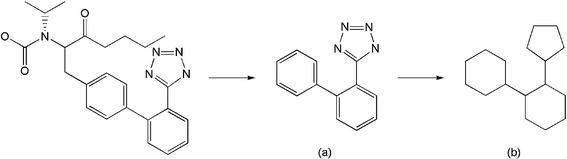 | ||
| Fig. 3 Hydrogen-free example of (a) atomic framework (or cyclic system) and (b) molecular framework (or skeletal cyclic system) of Diovan®. | ||
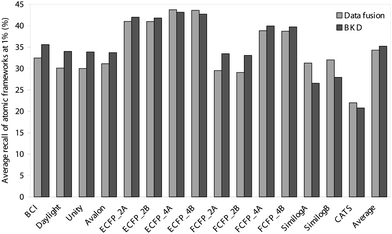 | ||
| Fig. 4 Comparison of the average percentage of atomic frameworks retrieved in the top 1% of the ranked test set obtained using BKD and data fusion. | ||
 | ||
| Fig. 5 Comparison of the average percentage of molecular frameworks retrieved in the top 1% of the ranked test set obtained using BKD and data fusion. | ||
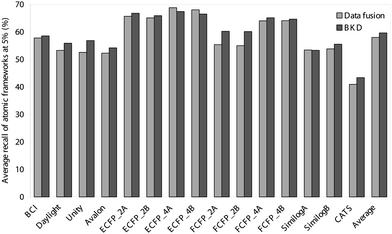 | ||
| Fig. 6 Comparison of the average percentage of atomic frameworks retrieved in the top 5% of the ranked test set obtained using BKD and data fusion. | ||
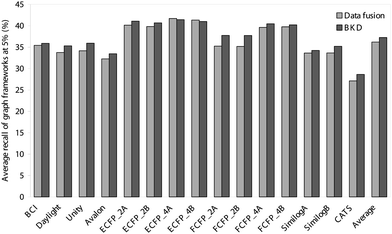 | ||
| Fig. 7 Comparison of the average percentage of molecular frameworks retrieved in the top 5% of the ranked test set obtained using BKD and data fusion. | ||
All the experiments carried out so far were performed using a version of the MDDR database in which every molecule was characterised by its neutral structure. However, drugs are used in vivo and further searches were hence carried out in order to see if any improvements in recall could be obtained by using the protonated states of the MDDR molecules. The pH component of Scitegic's Pipeline Pilot software35 was used to derive protonated molecular representations corresponding to a pH 6.8. However, very little difference was observed in the recalls obtained from the compounds in their protonated and neutral forms, with the latter normally being the more effective. There would hence appear to be little point in carrying out the additional processing required to produce the protonated representations.
The results presented here provide further evidence of the general effectiveness of the BKD and data fusion methods for virtual screening applications where multiple reference structures are available, and evidence of the general effectiveness of fingerprints based on 2D circular substructures, in particular the ECFP_4 fingerprints. If a single choice is required, then the best overall performance would seem to result from data fusion of the similarity scores of searches based on the ECFP_4B fingerprints. This is indicated as the combination of choice for several reasons. If we consider the choice of fingerprint first, then whilst the ECFP_4 descriptors achieved an excellent overall level of performance, they gave particularly good results when searching for structurally heterogeneous sets of molecules, a more challenging task than for highly self-similar sets of molecules. For this descriptor, the type-B binning scheme results in a very compact, reproducible representation that is only marginally inferior to the much larger, non-reproducible type-A binning scheme. Turning now to the search algorithms: data fusion is far less demanding of computational resources than is BKD and also does not require the specification of values for the latter's tuneable parameters; and an inspection of the standard deviations in Tables 1–4 shows that these tend to be larger (corresponding to a high level of variation in search performance) for BKD than for data fusion, suggesting a greater degree of consistency for the latter algorithm.
When considering the two search algorithms, it must be emphasised that we are dealing with a combination of characteristics, as evidenced by the fact that BKD does better than data fusion for some of the fingerprint types (e.g., Unity or Daylight): however, when used in combination with ECFP_4, the data fusion searches are to be preferred. It should also be emphasised that this preference for score-based data fusion over BKD is specific to the circumstances of these experiments, which involve just a limited number of active reference structures, as we have found that BKD is to be preferred when a proper training-set is available containing large numbers of both known actives and known inactives.36
Experimental
Our virtual screening system involves three main parts: a structural representation that is used to encode the molecules that are being searched; a searching method that ranks a database of molecules in order of decreasing probability of activity in response to a set of active reference structures; and a quantitative measure of the effectiveness of those rankings. The focus of this paper is the first of these factors, but it is appropriate to describe briefly the last two factors before discussing the many different types of 2D fingerprint that were evaluated.Searching algorithms
A previous study10 investigated a range of search algorithms that could be used when multiple reference structures were available. These experiments all involved a single type of fingerprint, specifically the Unity fingerprints produced by Tripos Inc.37 Two of the algorithms were found to provide a consistently high level of screening effectiveness: these algorithms were data fusion using the maximum of similarity scores and an approximate form of the BKD machine learning technique.Data fusion (or consensus scoring) involves combining the results of different similarity searches of a chemical database. Previous studies have involved the use of a single reference molecule, but characterised by several different representations or using several different similarity coefficients (see e.g. refs. 38, 39). An alternative approach, and the one used here, is to have a fixed representation and similarity coefficient, but to combine the search outputs obtained with several different reference structures. Assume that some database molecule i yields similarity scores of s1, s2...sn with the n different reference structures, then we have shown that effective searches are obtained by ranking the database molecules on the basis of the maximum of these scores, i.e., max{s1, s2...si...sn − 1, sn}; such searches are more effective than those resulting from the use of ranks, rather than scores, or the use of a fusion rule based on averaging.9, 10
The similarity scores were computed using the Tanimoto coefficient; for a molecule having a fingerprints with a bits set, of which c are also set in the fingerprint for a molecule that has b bits set, then the Tanimoto coefficient, Tc, is defined to be
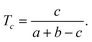
Some of the similarity scores necessitated the use of two non-binary similarity coefficients. Let xjA denote the occurrence of the j-th fragment (1 ≤
j
≤
n, the length of the integer vector) in molecule A (and similarly for molecule B). Then the similarity coefficients used were the non-binary form of the Tanimoto similarity coefficient3
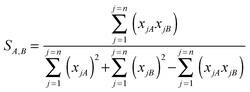
which is a Novartis coefficient that has been used in-house with the Similog descriptors.
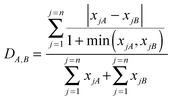
Binary kernel discrimination (BKD) is a machine learning technique that was first applied to virtual screening by Harper et al.40 The similarity between two compounds i and j, characterised by binary fingerprints of length M, that differ in dij positions, is computed by the kernel function Kλ(i,j),
| Kλ(i,j) = (λM−dij(1 − λ)dij)k/M |
being used to rank the molecules in the test-set, using the optimum values of λ and k found for the training set.36 When just a set of active reference structures is available, we have shown that the characteristics of the inactives can be approximated with a fair degree of accuracy by the characteristics of the entire database that is to be searched: a training-set can hence be generated by taking the set of reference structures and adding to it 100 molecules randomly selected from the database.10 The optimal values of λ and k were found to vary across the various types of fingerprint studied here, and extensive preliminary testing was required to identify the values that were used to obtain the main results that are discussed in the results and discussion section above. This variation in parameter value is a clear limitation of the BKD approach

Effectiveness of virtual screening
The experiments involved simulated virtual screening searches on the MDL Drug Data Report (MDDR) database.19 After removal of duplicates and molecules that could not be processed using local software, a total of 102![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 535 molecules was available for searching that were represented by each of the types of fingerprint described below. These molecules were searched using the eleven sets of active compounds that are listed in Table 5, which also contains the numbers of actives in each class and the numbers of active atomic and molecular frameworks (ring-system descriptors that are discussed above) in each class. The table also contains a numeric estimate of the level of structural diversity in each of the chosen sets of bioactives. The diversity estimate was obtained by matching each compound with every other in its activity class, calculating similarities using the Unity fingerprint and the Tanimoto coefficient and computing the mean of these intra-set similarities. The resulting similarity scores are listed in the right-hand part of Table 5, where it will be seen that the renin inhibitors are the most homogeneous and the cyclooxygenase inhibitors are the most heterogeneous.
535 molecules was available for searching that were represented by each of the types of fingerprint described below. These molecules were searched using the eleven sets of active compounds that are listed in Table 5, which also contains the numbers of actives in each class and the numbers of active atomic and molecular frameworks (ring-system descriptors that are discussed above) in each class. The table also contains a numeric estimate of the level of structural diversity in each of the chosen sets of bioactives. The diversity estimate was obtained by matching each compound with every other in its activity class, calculating similarities using the Unity fingerprint and the Tanimoto coefficient and computing the mean of these intra-set similarities. The resulting similarity scores are listed in the right-hand part of Table 5, where it will be seen that the renin inhibitors are the most homogeneous and the cyclooxygenase inhibitors are the most heterogeneous.
| Activity class | Number of | Similarity | |||
|---|---|---|---|---|---|
| Actives | Assemblies | Frameworks | Mean | SD | |
| 5HT3 antagonists | 752 | 438 | 237 | 0.35 | 0.12 |
| 5HT1A agonists | 827 | 478 | 271 | 0.34 | 0.10 |
| 5HT Reuptake inhibitors | 359 | 193 | 126 | 0.35 | 0.12 |
| D2 antagonists | 395 | 270 | 187 | 0.35 | 0.10 |
| Renin inhibitors | 1130 | 595 | 339 | 0.57 | 0.11 |
| Angiotensin II AT1 antagonists | 943 | 496 | 285 | 0.40 | 0.10 |
| Thrombin inhibitors | 803 | 451 | 295 | 0.42 | 0.13 |
| Substance P antagonists | 1246 | 633 | 380 | 0.40 | 0.11 |
| HIV protease inhibitors | 750 | 475 | 331 | 0.45 | 0.12 |
| Cyclooxygenase inhibitors | 636 | 308 | 139 | 0.27 | 0.09 |
| Protein kinase C inhibitors | 453 | 190 | 134 | 0.32 | 0.14 |
For each of the 11 activity classes, ten active compounds were selected for use as reference structures. The selections were done at random, subject to the constraint that no pair-wise similarity in a group exceeded 0.80 (using Unity fingerprints and the Tanimoto coefficient). The set of reference structures was searched against the MDDR database using the data fusion and BKD search algorithms described above, with the search being carried out once for each of the different types of fingerprint. The procedure was then repeated using ten different sets of reference structures, and in each search, a note was made of the recall, that is the percentage of the active molecules (i.e., those in the same class as those in the reference set) that occurred in the top 1% and the top 5% of the ranking resulting from that search. Formally, if a search retrieves the top x% of a ranked database, and this subset contains a of the A actives for that activity class, then the recall, Rx, is defined to be20
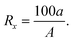
The results presented in Tables 1–4 are the mean and standard deviations for these recall values, averaged over each set of ten searches.
Fingerprint types
Having summarised the virtual screening environment and the two search algorithms, we now describe in some detail the four classes of fingerprint descriptors that we have studied. These are structural keys, hashed fingerprints, circular substructures and pharmacophores; in all, we evaluated ten different descriptors, of which seven are commercially available, two are used in-house at Novartis, and one was implemented from the literature description. Moreover, some of these descriptors were encoded in more than one way, to give a total of 15 types of fingerprint.Structural keys have been used in chemoinformatics for many years, and are usually encoded by a binary array, each element of which denotes the presence or absence of a specific 2D fragment. A predefined fragment dictionary lists the various fragment substructures that are encoded in the fingerprint. This study used the 1052 bit Barnard Chemical Information (BCI) fingerprints, which encode the following types of fragment substructure: augmented atoms, atom sequences, atom pairs, ring composition and ring fusion substructural fragments.41
Hashed fingerprints differ from structural keys in that they do not use a predefined dictionary. Instead, patterns are encoded in the fingerprint, where a pattern describes, for example, a path of length n bonds, i.e., (atom–bond–atom)n with the natures of the atoms and bonds defined. The set of patterns produced from any molecule of non-trivial size is obviously very large and differs from molecule to molecule. It is hence not possible to assign each potential pattern to a specific bit position in a fingerprint of predefined length; instead, the pattern is passed to a hashing function to generate a position (or positions) within the available length of the bit-string. The study used three different hashed fingerprints: 2048 bit Daylight fingerprints,42 988 bit Unity fingerprints37 and 2048 bit Avalon fingerprints. Daylight fingerprints encode each atom’s type, all augmented atoms and all paths of length 2–7 atoms. Unity fingerprints encode paths of length 2–6 atoms, and also include 60 structural keys for common atoms and ring counts. Avalon fingerprints are used for similarity search in Novartis' corporate data warehouse and encode atoms, augmented atoms, atom triplets and connection paths.
A circular substructure is a fragment descriptor where each atom is represented by a string of extended connectivity values that are calculated using a modification of the Morgan Algorithm.43 The study evaluated two different circular substructure descriptors from Scitegic's Pipeline Pilot Software:35 Extended Connectivity Fingerprints (ECFPs) and Functional Connectivity Fingerprints (FCFPs). The initial code assigned to an atom is based on the number of connections, the element type, the charge, and the mass for ECFPs and on six generalised atom-types (viz., hydrogen-bond donor, hydrogen-bond acceptor, positively ionisable, negatively ionisable, aromatic and halogen) for FCFPs. This code, in combination with the bond information and with the codes of its immediate neighbour atoms, is hashed to produce the next order code, which is mapped into an address space of size 232, and the process iterated until the required level of description has been achieved. The experiments here used the ECFP_2, ECFP_4, FCFP_2 and FCFP_4 fingerprints, where the numeric code denotes the diameter in bonds up to which features are generated.
The Scitegic software represents a molecule by a list of integers, each describing a molecular feature and each in the range −231 to 231. These integer lists were normalised in two ways, referred to as method A and method B. In method A, all the features present in the database were enumerated, so that each feature was given as its new code its rank in the sorted list of codes, with the length of the resulting fingerprints being the number of distinct features in the database. In method B, the integers describing a molecule were hashed to a bit-string of length 1024 bits. This inevitably means that collisions occur, with the result that method B loses some of the structural information that is retained by method A; however, the latter representation is dependent on the precise database that is being processed.
Pharmacophore points are features (such as a heteroatom or the centre of an aromatic ring) that are thought to be required for a molecule to show bioactivity. Pharmacophore fingerprinting involves generating all of the patterns of three or four pharmacophore points in a molecule, together with the corresponding inter-point distances, and then using the resulting 3D structural codes as descriptors for similarity searching or diversity analysis (see, e.g., refs. 17, 18, 30, 31). When used with 2D, rather than 3D, structural representations, the inter-atomic distances can be replaced by through-bond distances, and this approach forms the basis of the two pharmacophore fingerprints studied here: Similog keys9 and the Chemically Advanced Template Search (CATS) descriptor,28 both of which are based on generalised atom-types describing potential pharmacophores.
The Similog keys use a “DABE” atom-typing scheme based on the following four properties: hydrogen-bond donor (D), hydrogen-bond acceptor (A), bulkiness (B) and electropositivity (E). The presence or absence of these properties for an atom is encoded in a 4 bit string, and each triplet of atoms is represented by the three DABE strings and by the associated topological distances: in all, 8031 different codes were identified in the MDDR database. The Similog keys store the occurrence of each distinct code, and not just their presence or absence as in a conventional bit-string. A binning scheme was hence used to bin the occurrence data into 8 bit strings: the two binning schemes used (called method A and method B) are shown in Table 6.
The CATS descriptor is based on counts of atom-pair topological distances, with the following generalised types of atom being considered in the generation of the descriptor: lipophilic, positive, negative, hydrogen-bond donor and hydrogen-bond acceptor. The occurrences of the 15 possible pairs of pharmacophores are determined for distances up to 10 bonds to give a 150 element (i.e., 15 × 10) vector. The vectors were generated using the description in Fechner et al.29 and then converted to a binary fingerprint using method B in Table 6 (we only used method B for CATS as a substantial fraction of the keys occurred more than seven times in a molecule).
| Method A | Method B | 8 bit string |
|---|---|---|
| 1 occurrence | 20 ≤ occurrences < 21 | 10000000 |
| 2 occurrences | 21 ≤ occurrences < 22 | 11000000 |
| 3 occurrences | 22 ≤ occurrences < 23 | 11100000 |
| 4 occurrences | 23 ≤ occurrences < 24 | 11110000 |
| 5 occurrences | 24 ≤ occurrences < 25 | 11111000 |
| 6 occurrences | 25 ≤ occurrences < 26 | 11111100 |
| 7 occurrences | 26 ≤ occurrences < 27 | 11111110 |
| 8 and more occurrences | 27 ≤ occurrences | 11111111 |
Table 7 lists the abbreviated names that are used in the paper for each of the 15 types of fingerprints, where the A and B subscripts denote the type of normalisation scheme used for binning in the case of the ECFP, FCFP and Similog descriptors. The table also details statistical characteristics of each of these fingerprints: an inspection of the average numbers of bits and the densities (i.e., the mean number of bits that are set divided by the bit-string length and then expressed as a percentage) shows a very wide range of levels of molecular description.
| Name | Type | Normalised | Abbreviation | Length | Mean | SD | Max | Min | Density |
|---|---|---|---|---|---|---|---|---|---|
| Barnard Chemical Information | Dictionary-based | — | BCI | 1052 | 96.7 | 30.9 | 264 | 8 | 9.2 |
| Daylight | Hashed | — | Daylight | 2048 | 289.5 | 111.2 | 1046 | 24 | 14.1 |
| Unity | Hashed | — | Unity | 988 | 219.7 | 69.2 | 558 | 27 | 22.2 |
| Avalon | Hashed | — | Avalon | 2048 | 285.1 | 149.3 | 1076 | 16 | 13.9 |
| ECFP_2 | Circular substructure | A | ECFP_2A | 7445 | 32.4 | 9.4 | 103 | 5 | 0.4 |
| ECFP_2 | Circular substructure | B | ECFP_2B | 1024 | 31.8 | 9.1 | 98 | 5 | 3.1 |
| ECFP_4 | Circular substructure | A | ECFP_4A | 142864 | 54.0 | 17.0 | 191 | 8 | 0.0 |
| ECFP_4 | Circular substructure | B | ECFP_4B | 1024 | 52.4 | 16.0 | 177 | 8 | 5.1 |
| FCFP_2 | Circular substructure | A | FCFP_2A | 600 | 20.9 | 5.0 | 47 | 5 | 3.5 |
| FCFP_2 | Circular substructure | B | FCFP_2B | 1024 | 20.4 | 4.8 | 45 | 5 | 2.0 |
| FCFP_4 | Circular substructure | A | FCFP_4A | 30267 | 40.6 | 11.1 | 122 | 7 | 0.1 |
| FCFP_4 | Circular substructure | B | FCFP_4B | 1024 | 39.4 | 10.6 | 113 | 7 | 3.9 |
| Similog | Pharmacophore | A | SimilogA | 64248 | 1308.1 | 1437.2 | 14740 | 1 | 2.0 |
| Similog | Pharmacophore | B | SimilogB | 64248 | 863.5 | 900.1 | 10101 | 1 | 1.3 |
| CATS | Pharmacophore | — | CATS | 1200 | 96.0 | 36.2 | 453 | 1 | 8.0 |
Acknowledgements
We thank the following: Novartis Institutes for Biomedical Research for funding; MDL Information Systems Inc. for the provision of the MDDR database; and Barnard Chemical Information Ltd., Daylight Chemical Information Systems Inc., Pannanugget Consulting, the Royal Society, Tripos Inc. and the Wolfson Foundation for software and laboratory support; Dr Mark Johnson for access to ring-cycle software and Drs Bernd Rohde and Philippe Floersheim for helpful discussions and for access to the Avalon and Similog descriptors, respectively. The Krebs Institute for Biomolecular Research is a designated biomolecular sciences centre of the Biotechnology and Biological Sciences Research Council.References
- Virtual Screening for Bioactive Molecules, ed. H.-J. Böhm, and G. Schneider, Wiley-VCH, Weinheim, 2000 Search PubMed.
- Virtual Screening: an Alternative or Complement to High Throughput Screening?, ed. G. Klebe, Kluwer, Dordrecht, 2000 Search PubMed.
- P. Willett, J. M. Barnard and G. M. Downs, J. Chem. Inf. Comput. Sci., 1998, 38, 983–996 CrossRef CAS.
- R. P. Sheridan and S. K. Kearsley, Drug Discovery Today, 2002, 7, 903–911 CrossRef.
- Concepts and Applications of Molecular Similarity, ed. M. A. Johnson and G. M. Maggiora, Wiley, New York, 1990 Search PubMed.
- D. E. Patterson, R. D. Cramer, A. M. Ferguson, R. D. Clark and L. E. Weinberger, J. Med. Chem., 1996, 39, 3049–3059 CrossRef CAS.
- Y. C. Martin, J. L. Kofron and L. M. Traphagen, J. Med. Chem., 2002, 45, 4350–4358 CrossRef CAS.
- L. Xue, F. L. Stahura, J. W. Godden and J. Bajorath, J. Chem. Inf. Comput. Sci., 2001, 41, 746–753 CrossRef CAS.
- A. Schuffenhauer, P. Floersheim, P. Acklin and E. Jacoby, J. Chem. Inf. Comput. Sci., 2003, 43, 391–405 CrossRef CAS.
- J. Hert, P. Willett, D. J. Wilton, P. Acklin, K. Azzaoui, E. Jacoby and A. Schuffenhauer, J. Chem. Inf. Comput. Sci., 2004, 44, 1177–1185 CrossRef CAS.
- A. R. Leach and V. J. Gillet in An Introduction to Chemoinformatics, Kluwer, Dordrecht, 2003 Search PubMed.
- Handbook of Chemoinformatics ed. J. GasteigerWiley-VCH, Weinheim, 2003 Search PubMed.
- R. E. Carhart, D. H. Smith and R. Venkataraghavan, J. Chem. Inf. Comput. Sci., 1985, 25, 64–73 CrossRef CAS.
- P. Willett, V. Winterman and D. Bawden, J. Chem. Inf. Comput. Sci., 1986, 26, 36–41 CrossRef CAS.
- Combinatorial Library Design and Evaluation: Principles, Software Tools and Applications in Drug Discovery, ed. A. K. Ghose and V. N. Viswanadhan, Marcel Dekker, New York, 2001 Search PubMed.
- G. M. Downs and J. M. Barnard, Rev. Comput. Chem., 2002, 18, 1–40 Search PubMed.
- R. D. Brown and Y. C. Martin, J. Chem. Inf. Comput. Sci., 1996, 36, 572–584 CrossRef CAS.
- R. D. Brown and Y. C. Martin, J. Chem. Inf. Comput. Sci., 1997, 37, 1–9 CrossRef CAS.
- The MDL Drug Data Report database is available from MDL Information Systems Inc. at http://www.mdli.com.
- S. J. Edgar, J. D. Holliday and P. Willett, J. Mol. Graph. Modell., 2000, 18, 343–357 CrossRef CAS.
- J. E. Dubois, in Chemical Applications of Graph Theory, ed. A. T. Balaban, Academic Press, London, 1976, p. 161 Search PubMed.
- M. Randic, J. Chem. Inf. Comput.Sci., 1978, 18, 101–107 CrossRef CAS.
- P. Willett, J. Chem. Inf. Comput. Sci., 1979, 19, 159–162 CrossRef CAS.
- W. Schubert and I. Ugi, J. Am. Chem. Soc., 1978, 100, 37–41 CrossRef CAS.
- W. Bremser, Anal. Chim. Acta, 1978, 103, 355–365 CrossRef CAS.
- A. Bender, H. Y. Mussa, R. C. Glen and S. Reiling, J. Chem. Inf. Comput. Sci., 2004, 44, 170–178 CrossRef CAS.
- A. E. Klon, M. Glick, M. Thoma, P. Acklin and J. W. Davies, J. Med. Chem., 2004, 47, 2743–2749 CrossRef CAS.
- G. Schneider, W. Neidhart, T. Giller and G. Schmid, Angew. Chem., Int. Ed. Engl., 1999, 38, 2894–2896 CrossRef CAS.
- U. Fechner, F. Lutz, S. Renner, P. Schneider and G. Scheider, J. Comput.-Aided. Mol. Des., 2003, 17, 687–698 CrossRef CAS.
- H. Matter and T. Pötter, J. Chem. Inf. Comput. Sci., 1999, 39, 1211–1225 CrossRef CAS.
- J. S. Mason, I. Morize, P. R. Menard, D. L. Cheney, C. Hulme and R. F. Labaudiniere, J. Med. Chem., 1999, 42, 3251–3264 CrossRef CAS.
- G. W. Bemis and M. A. Murcko, J. Med. Chem., 1996, 39, 2887–2893 CrossRef CAS.
- Y. J. Xu and M. Johnson, J. Chem. Inf. Comput. Sci., 2001, 41, 181–185 CrossRef CAS.
- Y. J. Xu and M. Johnson, J. Chem. Inf. Comput. Sci., 2002, 42, 912–926 CrossRef CAS.
- Scitegic Inc. is at http://www.scitegic.com.
- D. J. Wilton, P. Willett, K. Lawson and G. Mullier, J. Chem. Inf. Comput. Sci., 2003, 43, 469–474 CrossRef CAS.
- Tripos Inc. is at http://www.tripos.com.
- C. M. R. Ginn, P. Willett and J. Bradshaw, Perspect. Drug Discovery Des., 2000, 20, 1–16 Search PubMed.
- N. Salim, J. D. Holliday and P. Willett, J. Chem. Inf. Comput. Sci., 2003, 43, 435–442 CrossRef CAS.
- G. Harper, J. Bradshaw, J. C. Gittins, D. V. S. Green and A. R. Leach, J. Chem. Inf. Comput. Sci., 2001, 41, 1295–1300 CrossRef CAS.
- Barnard Chemical Information Ltd. is at http://www.bci.gb.com/.
- Daylight Chemical Information Systems Inc. is at http://www.daylight.com.
- H. L. Morgan, J. Chem. Doc., 1965, 5, 107–113 Search PubMed.
Footnote |
| † This is one of a number of contributions on the theme of molecular informatics, published to coincide with the RSC Symposium “New Horizons in Molecular Informatics”, December 7th 2004, Cambridge UK. |
| This journal is © The Royal Society of Chemistry 2004 |