Convergent syntheses of polycyclic ethers. Illustrations of the utility of acetal-linked intermediates
Masayuki
Inoue
*
Department of Chemistry, and Research and Analytical Center for Giant Molecules, Graduate School of Science, Tohoku University, Sendai 980-8578, Japan. E-mail: inoue@ykbsc.chem.tohoku.ac.jp
First published on 14th June 2004
Abstract
One of the most characteristic and spectacular class of compounds isolated from marine sources is the polycyclic ethers. Following the initial report of the structural determination of brevetoxin B, a variety of novel polycyclic ethers have begun to surface. These natural products include ciguatoxins, gambierol, gambieric acids, yessotoxins and gymnocins, each of which exhibit distinct biological properties such as cytotoxicity and neurotoxicity, as well as antiviral and antifungal activities. Because of these intriguing biological activities and their complex molecular architecture, the total synthesis of these compounds has been pursued by many laboratories over two decades. In particular, the development of novel convergent strategies to assemble the structural fragments is crucial for the successful construction of these nano-scale molecules. This Perspective will focus on a recent convergent methodology using an acetal-linkage as a key motif. Application of this methodology culminated in the total syntheses of gambierol and ciguatoxin CTX3C.
 Masayuki Inoue | Masayuki Inoue was born in Tokyo in 1971. He received a BSc degree in Chemistry from the University of Tokyo in 1993. In 1998, he obtained his PhD from the same university, working under the supervision of Professors K. Tachibana and M. Sasaki, on synthetic studies of ciguatoxin. After spending highly rewarding years with Professor S. J. Danishefsky at the Sloan-Kettering Institute for Cancer Research (1998–2000), working on the total synthesis of frondosin B and illicinones, he joined the Graduate School of Science at Tohoku University as an assistant professor of Professor Hirama's group. At Tohoku University, he was promoted to lecturer in 2003, and then to associate professor in 2004. There, he completed the total synthesis of ciguatoxin CTX3C, TMC-95A, and merrilactone A. He received both the Young Scientist's Research Award in Natural Product Chemistry and the Chugai Award in Synthetic Organic Chemistry in 2001. His research interests include the synthesis, design and study of biologically important molecules, with a particular emphasis on the total synthesis of structurally complex natural products. |
Introduction
Since brevetoxin B (Fig. 1, 1) was shown to have an unprecedented polycyclic ether structure by X-ray crystallography in 1981,1 various natural products with similar skeletons have been structurally elucidated using modern spectroscopic techniques.2 These molecules are made up of a single carbon chain locked into a semi-rigid ladder-like structure. The striking regularity with which the oxygen atoms bridge the polycyclic framework and the all-trans/syn/trans ring fusions are remarkable features of these molecules. Despite this common polycyclic motif, they show diverse biological activities with extreme potency. | ||
| Fig. 1 Structure of representative polycyclic ethers. | ||
Brevetoxins (1, 23) are potent ichthyotoxins (2: LC100 4 ng mL−1 to guppies), and were isolated from the dinoflagellate Gymnodinium breve, a causative organism of the massive “red tide” fish kills. A similar red tide dinoflagellate Gymnodiniummikimotoi produces gymnocin-A (3).4 In contrast to brevetoxins, 3 is weakly toxic in fish, but is cytotoxic to P388 mouse leukemia cells.4 Ciguatoxins (5,566), potent neurotoxins, were isolated as the toxic principles of the wide-spread seafood poisoning known as ciguatera.7 These toxins accumulate in fish though the food chain, starting from the dinoflagellate Gambierdiscus toxicus.8 Some strains of this organism produce not only ciguatoxins, but also other polycyclic ethers, such as gambierol (4), that exhibit toxicity against mice.9
The novel functions of these molecules have attracted interest from both the biological and chemical communities, and many investigations have focused on elucidating their biological targets. However, receptor proteins have only been identified for brevetoxins and ciguatoxins.10,11 These molecules have been shown to bind strongly with the voltage sensitive sodium channels of excitable membranes, causing them to open, thereby allowing sodium ion influx.
Over the past two decades, a number of laboratories have attempted the chemical construction of these compounds due to their unusual molecular architecture, biological activity and association with important phenomena such as red tide and food poisonings. Their exquisitely complex structures have served as inspiration for the development of new methodologies in organic synthesis, and as an elegant platform for exhibiting the creativity of modern organic chemists.12 In 1995, Nicolaou, a pioneer in the field of polyether syntheses, reported the first total synthesis of brevetoxin B (1).13 This major advance was followed by the synthesis of brevetoxin A (2) by the same laboratory in 1998.14 In the last five years, even more rapid progress has been made by many laboratories. The efforts culminated in the total syntheses of ciguatoxin CTX3C (5) by Hirama et al. in 2001,15,16 gambierol (4) by both Sasaki17 and Kadota and Yamamoto18 in 2002, brevetoxin B (1) by Nakata in 2002,19 and gymnocin-A (3) by Sasaki in 2003.20 These landmark achievements were made possible by the development of a number of important reactions and methodologies.
The large and complex structures of these molecules necessitate a highly efficient synthetic strategy with excellent material throughput and minimum synthetic transformations. Because linear construction of the ether rings is virtually impossible due to the size of the molecule, development of an efficient methodology for coupling the fragments, suitable for use in the latter stages of synthesis, has been particularly important for the total synthesis. Various convergent strategies have been published, and successfully applied to syntheses of natural products and their structural fragments. Among these, this Perspective will focus on a recent methodology using an acetal-linkage as a key motif. Development of such protocols and their application to the total synthesis of gambierol (4) and CTX3C (5) will also be discussed in this Perspective.
Convergent synthetic strategy via acetal-linked intermediate
The generalized route to the fused ether array through the acetal-linked intermediate is illustrated in Scheme 1. Firstly, acetal 9 is synthesized through coupling of the synthetic fragments 7 and 8. Construction of the ether ring from 9via intramolecular C–C bond formation would then give O-linked oxacycle 12, of which cyclization via another C–C bond formation would afford fused ether array 13. Formidable challenges lie in the construction of the first ring with the introduction of the two stereocenters of 12 and efficient cyclization of the second medium-sized ether ring (12 → 13). Generally, construction of medium ring structures has been hampered by difficulties in effecting ring closure due to unfavorable entropy factors as well as transannular nonbonding interactions. One of the advantages of this strategy is that the carbon chains of O-linked oxacycle 12 are in close proximity in the global minimum conformer, as indicated by molecular modeling studies of 14 (Fig. 2), and thus 12 has the appropriate conformation for cyclization through C–C bond formation. | ||
| Scheme 1 Convergent strategy via an acetal-linked intermediate. | ||
 | ||
| Fig. 2 Minimized structure of O-linked oxacycle 14 (MacroModel Ver. 8.0, MM2*). | ||
There are two possible reaction courses for the first intramolecular ether ring formation (Scheme 1): (i) nucleophilic addition to the acetal carbon (10 → 12) and (ii) addition of the α-oxyradical to the olefin (11 → 12). In both cases, development of stereoselective methods to control the two bridgehead stereocenters is the critical issue, and will be the particular focus of this Perspective.
Nucleophilic addition of γ-alkoxyallylsilane to cyclic acetal
In the early 1990s, Yamamoto's group published a series of papers on the intramolecular Lewis acid-mediated reaction of γ-alkoxyallylmetals with acetals to obtain cyclic ethers (15 → 16 or 17, Fig. 3).21 This pioneering work was extended to convergent methodologies to assemble polycyclic ethers by Martín22 and Tachibana.23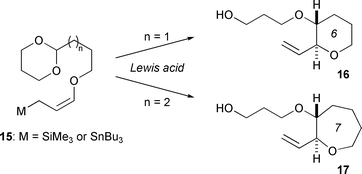 | ||
| Fig. 3 Intramolecular cyclization of γ-alkoxyallylmetal with acetal developed by Yamamoto.21 | ||
In studies toward the total synthesis of ciguatoxin, Tachibana and co-workers developed a method for the synthesis of 6/9/6-tricyclic system in 1997.23 Their synthesis started with coupling between diol 18 and aldehyde 19 by acetalization (Scheme 2). Lithiation of allylic ether 20 with s-BuLi and in situ trapping of the resulting anion with either n-Bu3SnCl or Me3SiCl gave γ-alkoxyallylmetal 21 or 22. Interestingly, Lewis acids cleaved the same less-hindered C–O acetal bonds of 21 and 22, but generated different stereoisomers. Whereas cyclization of allylstannane 21 exclusively formed the undesired trans/anti/trans-isomer 23, allylsilane 22 led to selective formation of the desired trans/syn/trans-isomer 14.
 | ||
| Scheme 2 Tachibana's construction of O-linked oxacycle through γ-alkoxyallylmetal-acetal cyclization (1997).23 | ||
The mechanistic rationale for these cyclizations is shown in Scheme 2. The reactive allylstannane in 21 would react in an SN2-like manner; cleavage of the C–O bond of acetal would take place simultaneously with the C–C bond formation between the γ-allylic and acetal carbons. Therefore, cyclization would selectively produce the undesired 23. On the other hand, it was assumed that alternative use of the significantly less reactive γ-alkoxyallylsilane 2224 would enable the reaction to take place via an SN1-like transition state. In this pathway, the memory of chirality of the acetal carbon would disappear to give an oxonium cation, which would react with allylsilane in order to minimize unfavorable steric interactions to afford the desired isomer 14.
Compound 14 was then converted to α-bromoketone 25 (Scheme 3), which was smoothly cyclized to 9-membered ring 26 by the action of SmI2.25 Thus, O-linked oxacycle 25 proved to be suitable for medium-sized ether ring formation as indicated by the molecular mechanics in Fig. 2. Finally, the targeted 6/9/6-ring system 27 was synthesized from 26 in several functional group manipulations.
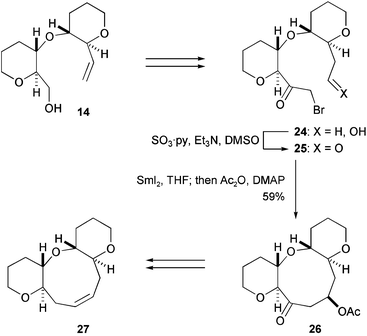 | ||
| Scheme 3 Tachibana's synthesis of 6/9/6-tricyclic ring system (1997).23 | ||
Sasaki and Tachibana exploited this strategy for the synthesis of decacyclic ciguatoxin model 34 in 1998 (Scheme 4).26 Their preliminary model experiments revealed that the seven-membered acetal was more reactive toward intramolecular nucleophilic addition than the six-membered counterpart.27 Thus, 1,4-diol 29 was selected as a structural fragment in anticipation of the high yield and diastereoselectivity of Lewis acid-mediated cyclization. Acetal formation between β-alkoxy aldehyde 28 and 1,4-diol 29 was realized in the presence of a catalytic amount of Sc(OTf)328 The reaction took place at room temperature and produced seven-membered acetal 30 in 90% yield. Lithiation of 30 followed by treatment with Et3SiCl gave γ-alkoxyallylsilane 31. Upon treatment of 31 with TiCl4–PPh3, the desired O-linked 32 was obtained as a major product in 36% yield, together with three other stereoisomers. Similarly to the synthesis of the previous compound, 32 was converted to 6/9/7/6-ring system 33, which was further converted to decacyclic ciguatoxin model 34 through coupling with the JKLM ring fragment. These studies clearly demonstrated that the acetal-based convergent strategy provided a general and powerful method for the construction of complex trans-fused ether polycycles. Furthermore, tuning the reactivity of both the acetals and nucleophiles was shown to be important in obtaining O-linked oxacycles with the desired stereochemistries.
 | ||
| Scheme 4 Synthesis of decacyclic ciguatoxin model by Sasaki and Tachibana (1998).26 | ||
Nucleophilic addition of γ-alkoxyallylstannane to α-acetoxy ether
In 2001, Kadota and Yamamoto developed a stereoselective approach to polycyclic ring systems using the intramolecular cyclization of γ-alkoxyallylstannanes to α-acetoxy ethers (Scheme 5).29 Carboxylic acid 35 and alcohol 36 were coupled through esterification to afford 37. After deprotection of the silyloxy group of 37, the alcohol was converted to allylstannane 39via38. α-Acetoxy ether 40 was then prepared from ester 39 according to Rychnovsky protocol:30 (i) reduction of 39 with DIBAL and (ii) in situ trapping of hemiacetal with acetic anhydride. Treatment of mixed acetal 40 with MgBr2·OEt2 resulted in the intramolecular cyclization to give seven-membered ring 41 as an exclusive isomer (>95 : 5), which was transformed to 6/7/7/6-ring system 43 by the ring-closing olefin metathesis reaction (RCM).31 | ||
| Scheme 5 Yamamoto's intramolecular cyclization of γ-alkoxyallylstannane with α-acetoxy ether (2001).29 | ||
The stereochemical outcome of the reaction of allylstannane 40 did not reflect the chirality at the α-acetoxy group, presumably because of its facile formation of the oxonium cation in contrast to cyclic acetal 21 in Scheme 2. Thus, the observed stereoselectivity can be explained by the SN1-like transition state model. The allylic stannane moiety is oriented to a pseudo-equatorial position in order to avoid 1,3-diaxial repulsion, and the oxonium cation moiety bearing a substituted tetrahydropyranyl group R also prefers a pseudo-equatorial position as depicted by the transition state structure 45, which leads to 41.
Kadota et al. accomplished the highly convergent total synthesis of gambierol (4) by exploiting newly developed technology (Scheme 6).18 ABC-segment 46 and FGH segment 47 were condensed, and then a series of reactions including desilylation, attachment of allylic stannane moiety and subsequent Rychnovsky protocol gave α-acetoxy ether 48. In contrast to the model experiments, treatment of 48 with MgBr2·OEt2 afforded the undesired stereoisomer 51 as the major component. Interestingly, BF3·Et2O-mediated cyclization of α-chloroacetyl 49 led to the selective formation of 50. This improved diastereoselectivity is presumably due to more facile formation of the oxonium cation intermediate from the chloroacetyl group than from the acetyl group.
 | ||
| Scheme 6 Total synthesis of gambierol by Kadota et al. (2002).18 | ||
The obtained diene 50 was subjected to RCM, leading to octacyclic gambierol skeleton 53. Finally, 53 was successfully converted to the natural product 4 through functional group modification, side chain introduction and final deprotection. It should be emphasized that the use of an esterification reaction for segment coupling and the seven-step construction of the two fused ring system makes the present methodology highly efficient and practical.
Intramolecular radical cyclization from mixed-acetal
In 1998, Sasaki and Tachibana reported the convergent synthesis of O-linked oxepane based on the intramolecular radical reaction (Scheme 7).32 Acetalization between 54 and 55 led to six-membered acetal 56, which was converted to O,Se-acetal 57 through the regioselective cleavage of the less hindered C–O bond using i-Bu2AlSePh.33 Protection of the primary hydroxyl group in 57, subsequent removal of the silyl group and attachment of β-(E)-alkoxyacrylate afforded 58. Treatment of 58 with n-Bu3SnH in the presence of Et3B34 resulted in formation of the desired O-linked oxepane 61 as a single diastereomer in high yield. | ||
| Scheme 7 Intramolecular radical cyclization developed by Sasaki (1998).32 | ||
The stereoselectivity of the cyclization is explained as indicated in Scheme 7. Initially, the stereochemical information of the acetal carbon was lost upon formation of the radical intermediate. The β-alkoxyacrylate favored the extended s-trans- over s-cis-conformation in order to avoid the 1,3-diaxial-like interactions.35 Furthermore, the steric interactions between the bulky alkoxy group and the s-trans-alkoxyacrylate of the pseudo-equatorial 59 resulted in preference for the pseudo-axial 60, from which the desired isomer 61 was the only possible outcome among the four possible isomers.
This remarkable protocol based on radical cyclization was further modified and refined by Hirama's laboratory, which culminated in their total synthesis of ciguatoxin CTX3C (5, Scheme 8).15,16 Sc(OTf)3-promoted coupling of diol 62 and aldehyde 63 delivered seven-membered acetal 64. The seven-membered acetal was selected over the six-membered counterpart based on their model experiments, which again indicated the superior reactivity of the seven-membered acetal.36 Indeed, the acetal cleavage reaction of 64 was realized using Me3SiOTf and Me3SiSPh,37 which led to O,S-acetal 65 without affecting the potentially reactive C49-spiroacetal. After three synthetic steps from 65, β-alkoxyacrylate 67 was then subjected to radical cyclization using n-Bu3SnH and 2,2′-azobisisobutyronitrile (AIBN), giving rise to the desired oxepane 68 as the sole isomer. For this step, the generated C27-radical added to the α,β-unsaturated ester in a completely stereo- and chemoselective manner. To prepare for cyclization of the last remaining F-ring, the carbon chains of 68 were transformed to the terminal olefins of 69 in six steps. The RCM reaction of 69 using Grubbs catalyst 42 provided the protected CTX3C 70, which was converted to the target CTX3C (5) through global deprotection. Hence, this total synthesis of CTX3C proved the power and reliability of the O,S-acetal strategy to build complex polyether structures.
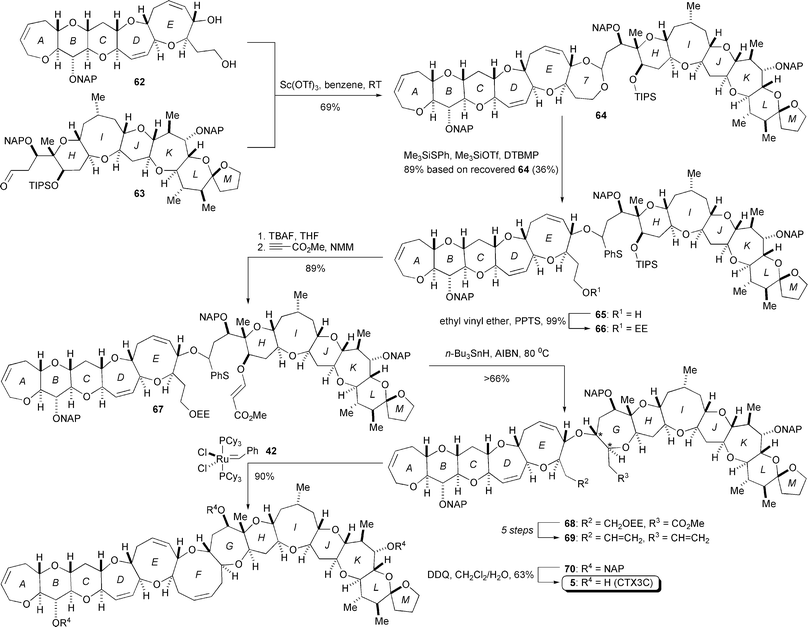 | ||
| Scheme 8 Total synthesis of ciguatoxin CTX3C by Hirama (2001 and 2002).15,16 | ||
In 2004, Inoue and Hirama reported their second-generation total synthesis of CTX3C (5), which is more concise, efficient and applicable (Scheme 9).38 The alternative synthetic strategy relied on the direct construction of the key O,S-acetal 74 under mild conditions.39 The α-chloride was installed on to right wing sulfide 71 using NCS, leading to α-chlorosulfide 72. The obtained 72 and left wing alcohol 73 were coupled by the action of AgOTf in the presence of 2,6-di-tert-butyl-4-methylpyridine (DTBMP) and 4 Å MS.40 In this way, O,S-acetal 73 was obtained in high yield, thus accomplishing direct construction of the key intermediate. The F- and G-rings were then constructed from 73 similarly to the first-generation synthesis, which resulted in their second-generation total synthesis of 5. Importantly, halophilic silver salt, used in the coupling, is highly chemoselective, and allows the use of various functional groups, and this method installed two rings in only eight synthetic transformations.
 | ||
| Scheme 9 Second generation total synthesis of CTX3C by Inoue and Hirama (2004).38 | ||
Conclusion and perspectives
As a result of rapid progress over the last five years, convergent strategies utilizing acetal-linked intermediates have been improved and polished up to a satisfactory level in terms of overall stereoselectivity, efficiency and practicability. A number of the developed synthetic technologies are at the cutting edge of contemporary organic chemistry, particularly the various methods used to prepare key acetal-linked intermediates, the subsequent highly stereoselective constructions of the ether rings, and the RCM reaction to build medium-sized ethers from O-linked oxacycles. Importantly, these reaction sequences only require 7–8 synthetic transformations to construct two new rings, and proved to be highly applicable to complex polycyclic structures, such as gambierol and ciguatoxin CTX3C. It is anticipated that the materials generated through these total syntheses will help to better understand their detailed biological mechanisms of action.However, de novo chemical synthesis of polycyclic ethers has not yet become a routine preparative method to obtain natural products and their analogs. The existing total syntheses typically require more than 100 total steps. Although such syntheses represent impressive contributions to organic chemistry, development of even more practical and general synthetic routes remains a key challenge for the future.
Notes and references
- Y.-Y. Lin, M. Risk, S. M. Ray, D. Van Engen, J. Clardy, J. Golik, J. C. James and K. Nakanishi, J. Am. Chem. Soc., 1981, 103, 6773 CrossRef CAS.
- For reviews, see: (a) T. Yasumoto and M. Murata, Chem. Rev., 1993, 93, 1897 CrossRef CAS; (b) T. Yasumoto, Chem. Rec., 2001, 1, 228 Search PubMed.
- Y. Shimizu, H.-N. Chou, H. Bando, G. Van Duyne and J. C. Clardy, J. Am. Chem. Soc., 1986, 108, 514 CrossRef CAS.
- M. Satake, M. Shoji, Y. Oshima, H. Naoki, T. Fujita and T. Yasumoto, Tetrahedron Lett., 2002, 43, 5829 CrossRef CAS.
- M. Satake, M. Murata and T. Yasumoto, Tetrahedron Lett., 1993, 34, 1975 CrossRef CAS.
- M. Murata, A.-M. Legrand, Y. Ishibashi, M. Fukui and T. Yasumoto, J. Am. Chem. Soc., 1990, 112, 4380 CrossRef CAS.
- (a) P. J. Scheuer, Tetrahedron, 1994, 50, 3 CrossRef CAS; (b) R. J. Lewis, Toxicon, 2001, 39, 97 CrossRef CAS.
- T. Yasumoto, I. Nakajima, R. Bagnis and R. Adachi, Nippon Suisan Gakkaishi, 1977, 43, 1015 Search PubMed.
- (a) M. Satake, M. Murata and T. Yasumoto, J. Am. Chem. Soc., 1993, 115, 361 CrossRef CAS; (b) A. Morohashi, M. Satake and T. Yasumoto, Tetrahedron Lett., 1998, 39, 97 CrossRef CAS.
- (a) M. A. Poli, T. J. Mende and D. G. Baden, Mol. Pharmacol., 1986, 30, 129 Search PubMed; (b) A. Lombet, J.-N. Bidard and M. Lazdunski, FEBS Lett., 1987, 219, 355 CrossRef CAS.
- Recent examples with insight into polyether activities: M. Inoue, M. Hirama, M. Satake, K. Sugiyama and T. Yasumoto, Toxicon, 2003, 41, 469 Search PubMed.
- For recent reviews, see: (a) E. Alvarez, M.-L. Candenas, R. Pérez, J. L. Ravelo and J. D. Martín, Chem. Rev., 1995, 95, 1953 CrossRef CAS; (b) Y. Mori, Chem. Eur. J., 1997, 3, 849 CAS; (c) F. P. Marmsäter and F. G. West, Chem. Eur. J., 2002, 8, 4347; (d) P. A. Evans and B. Delouvrie, Curr. Opin. Drug Discovery Dev., 2002, 5, 986 Search PubMed.
- (a) K. C. Nicolaou, F. P. J. T. Rutjes, E. A. Theodorakis, J. Tiebes, M. Sato and E. Untersteller, J. Am. Chem. Soc., 1995, 117, 1173 CrossRef CAS; (b) K. C. Nicolaou, F. P. J. T. Rutjes, E. A. Theodorakis, J. Tiebes, M. Sato and E. Untersteller, J. Am. Chem. Soc., 1995, 117, 10252 CrossRef CAS.
- (a) K. C. Nicolaou, Z. Yang, G. Shi, J. L. Gunzner, K. A. Agrios and P. Gärtner, Nature, 1998, 392, 264 CrossRef CAS; (b) K. C. Nicolaou, J. L. Gunzner, G. Shi, K. A. Agrios, P. Gärtner and Z. Yang, Chem. Eur. J., 1999, 5, 646 CrossRef CAS.
- M. Hirama, T. Oishi, H. Uehara, M. Inoue, M. Maruyama, H. Oguri and M. Satake, Science, 2001, 294, 1904 CrossRef CAS.
- (a) M. Inoue, H. Uehara, M. Maruyama and M. Hirama, Org. Lett., 2002, 4, 4551 CrossRef CAS; (b) For an account, see: M. Inoue and M. Hirama, Synlett, 2004, 577 Search PubMed.
- (a) H. Fuwa, M. Sasaki, M. Satake and K. Tachibana, Org. Lett., 2002, 4, 2981 CrossRef CAS; (b) H. Fuwa, N. Kainuma, K. Tachibana and M. Sasaki, J. Am. Chem. Soc., 2002, 124, 14983 CrossRef CAS.
- (a) I. Kadota, H. Takamura, K. Sato, A. Ohno, K. Matsuda and Y. Yamamoto, J. Am. Chem. Soc., 2003, 125, 46 CrossRef CAS; (b) I. Kadota, H. Takamura, K. Sato, A. Ohno, K. Matsuda, M. Satake and Y. Yamamoto, J. Am. Chem. Soc., 2003, 125, 11893 CrossRef CAS.
- G. Matsuo, K. Kawamura, N. Hori, H. Matsukura and T. Nakata, Abstracts of 44th Symposium on the Chemistry of Natural Products, Tokyo, Japan, 2002, p 163 Search PubMed.
- C. Tsukano and M. Sasaki, J. Am. Chem. Soc., 2003, 125, 14294 CrossRef CAS.
- (a) J.-i. Yamada, T. Asano, I. Kadota and Y. Yamamoto, J. Org. Chem., 1990, 55, 6066 CrossRef CAS; (b) I. Kadota, V. Gevorgyan, J. Yamada and Y. Yamamoto, Synlett, 1991, 823 CrossRef CAS; (c) I. Kadota, K. Miura and Y. Yamamoto, J. Chem. Soc., Chem. Commun., 1994, 1953 RSC.
- E. Alvarez, M. T. Díaz, L. Hanxing and J. D. Martín, J. Am. Chem. Soc., 1995, 117, 1437 CrossRef CAS.
- (a) M. Inoue, M. Sasaki and K. Tachibana, Tetrahedron Lett., 1997, 38, 1611 CrossRef CAS; (b) M. Inoue, M. Sasaki and K. Tachibana, Tetrahedron, 1999, 55, 10949 CrossRef CAS.
- In general, addition of allyltrimethylsilane to various electrophiles has been shown to be 103–104 times slower than that of allyltributylstannane. H. Mayr and M. Patz, Angew. Chem., Int. Ed. Engl., 1994, 33, 938 Search PubMed.
- (a) J. Inanaga, Y. Yokoyama, Y. Honda and M. Yamaguchi, Tetrahedron Lett., 1991, 32, 6371 CrossRef CAS; (b) G. Molander and C. R. Harris, Chem. Rev., 1996, 96, 307 CrossRef CAS.
- (a) M. Inoue, M. Sasaki and K. Tachibana, Angew. Chem., Int. Ed. Engl., 1998, 37, 965 CrossRef CAS; (b) M. Inoue, M. Sasaki and K. Tachibana, J. Org. Chem., 1999, 64, 9416 CrossRef CAS.
- Very recently, convergent synthesis of a tetracyclic ether system via α-cyano ether was reported by Oishi, which also utilized a seven-membered acetal as a key intermediate. T. Oishi, K. Watanabe and M. Murata, Tetrahedron Lett., 2003, 44, 7315 Search PubMed.
- (a) S. Fukuzawa, T. Tsushimoto, T. Hotaka and T. Hiyama, Synlett, 1995, 1077 CrossRef CAS; (b) Y. Ishihara, Y. Karumi, M. Kubota and H. Yamamoto, Synlett, 1996, 839 CrossRef CAS.
- (a) I. Kadota, A. Ohno, K. Matsuda and Y. Yamamoto, J. Am. Chem. Soc., 2001, 123, 6702 CrossRef CAS; (b) I. Kadota, A. Ohno, K. Matsuda and Y. Yamamoto, J. Am. Chem. Soc., 2002, 124, 3562 CrossRef CAS.
- (a) V. H. Dahanukar and S. D. Rychnovsky, J. Org. Chem., 1996, 61, 8317 CrossRef CAS; (b) D. J. Kopecky and S. D. Rychnovsky, J. Org. Chem., 2000, 63, 191 CrossRef.
- For excellent reviews, see: (a) T. M. Trnka and R. H. Grubbs, Acc. Chem. Res., 2001, 34, 18 CrossRef CAS; (b) A. Fürstner, Angew. Chem., Int. Ed., 2000, 39, 3012 CrossRef CAS.
- (a) M. Sasaki, M. Inoue, T. Noguchi, A. Takeichi and K. Tachibana, Tetrahedron Lett., 1998, 39, 2783 CrossRef CAS; (b) M. Sasaki, T. Noguchi and K. Tachibana, Tetrahedron Lett., 1999, 40, 1337 CrossRef CAS; (c) M. Sasaki, T. Noguchi and K. Tachibana, J. Org. Chem., 2002, 67, 3301 CrossRef CAS.
- K. Maruoka, T. Miyazaki, M. Ando, Y. Matsumura, S. Sakane, K. Hattori and H. Yamamoto, J. Am. Chem. Soc., 1983, 105, 2831 CrossRef CAS.
- K. Nozaki, K. Oshima and K. Utimoto, Bull. Chem. Soc. Jpn., 1990, 63, 2578 CAS.
- E. Lee, J. S. Tae, C. Lee and C. M. Park, Tetrahedron Lett., 1993, 34, 4831 CrossRef CAS.
- H. Imai, H. Uehara, M. Inoue, H. Oguri, T. Oishi and M. Hirama, Tetrahedron Lett., 2001, 42, 6219 CrossRef CAS.
- S. Kim, J. Y. Do, S. H. Kim and D. Kim, J. Chem. Soc., Perkin Trans. 1, 1994, 2357 RSC.
- M. Inoue, K. Miyazaki, H. Uehara, M. Maruyama and M. Hirama, Proc. Natl. Acad. Sci. U.S.A, in press Search PubMed.
- (a) M. Inoue, G. X. Wang, J. Wang and M. Hirama, Org. Lett., 2002, 4, 3439 CrossRef CAS; (b) M. Inoue, J. Wang, G. X. Wang, Y. Ogasawara and M. Hirama, Tetrahedron, 2003, 59, 5645 CrossRef CAS; (c) M. Inoue, S. Yamashita and M. Hirama, Tetrahedron Lett., 2004, 45, 2053 CrossRef CAS.
- J. C. McAuliffe and O. Hindsgaul, Synlett, 1998, 307 CAS.
| This journal is © The Royal Society of Chemistry 2004 |