A one-pot remote allylic hydroxylation and Baeyer–Villiger oxidation of a bicyclo[3.2.0]hept-2-en-6-one by Cunninghamella echinulata NRRL 3655
Ian J. S.
Fairlamb
*a,
Stephanie
Grant
a,
Gideon
Grogan
*b,
David A.
Maddrell
b and
Josephine C.
Nichols
b
aDepartment of Chemistry, University of York, Heslington, York, UK YO10 5DD. E-mail: ijsf1@york.ac.uk; Fax: 44 1904 432516; Tel: 44 1904 434091
bYork Structural Biology Laboratory, Department of Chemistry, University of York, Heslington, York, UK YO10 5YW. E-mail: gg12@york.ac.uk
First published on 10th June 2004
Abstract
7-exo-Methyl-7-endo-phenylbicyclo[3.2.0]hept-2-en-6-one 3 undergoes Baeyer–Villiger and allylic oxidation, to yield novel hydroxylactone 8 in good yield by Cunninghamella echinulata NRRL 3655, representing a one step biocatalytic access to a cyclopentanoid scaffold with three chiral centers. Interesting, allylic oxidation occurs with transposition of the double bond.
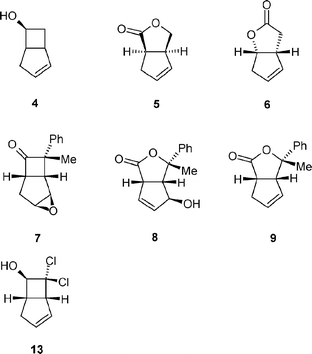
Whole cell preparations of filamentous fungi have been used widely since the 1950s as biocatalysts often because of superior regio- and enantioselectivities observed in a range of oxidation reactions, compared to abiotic chemical methods. In addition to hydroxylation1 and N-2 and O-demethylation3 reactions, organisms such as Beauveria bassiana4 and Cunninghamella echinulata have been used to catalyse preparatively useful Baeyer–Villiger reactions.5Cunninghamella echinulata NRRL 3655 has proved most useful in the latter case, particularly in the transformation of bicyclo[3.2.0]hept-2-en-6-one 1,6 derivatives of which can serve as important synthons in the preparation of prostanoids7 and pheromones.8 The interest in this approach lies in the multifunctional potential of the bicyclic heptanone nucleus, which can, after enantioselective biological Baeyer–Villiger oxidation and hydrolysis, yield chiral hydroxy acids and cyclopentanoid intermediates with two new chiral centers. In the wake of current interest in the one step preparation of multifunctionalised chiral cyclopentanoid scaffolds,9 we were interested to use biological Baeyer–Villiger reactions as a means of resolving racemic mixtures of bicyclo[3.2.0]hept-2-en-6-one derivatives such as 1 (Fig. 1). The resolved ketones would serve as intermediates in our syntheses of a range of ligands for palladium complexes.10Cunninghamella echinulata NRRL 3655 was the catalyst of choice as it had been reported to catalyse the apparent resolution of bicyclo[3.2.0]hept-2-en-6-one to yield the 3-oxa lactone 5 exclusively,6 in contrast to the bacterial Baeyer–Villiger biocatalysts, noted for their ability to catalyse the enantiodivergent oxidative biotransformation of this ketone series to yield approximately equimolar quantities of optically active 2- and 3-oxa lactones (5 and 6).11 In this paper, in addition to well-documented reductions and lactone formations of the bicyclo[3.2.0]hept-2-en-6-one system, we have unearthed a surprising and high yielding double oxidation of 7-exo-methyl-7-endo-phenylbicyclo[3.2.0]hept-2-en-6-one 3, wherein both Baeyer–Villiger oxidation and the formation of an allylic alcohol in the cyclopentene ring have occurred both regio- and stereo-selectively.
![7,7-Disubstituted bicyclo[3.2.0]hept-2-en-6-ones.](/image/article/2004/OB/b406016d/b406016d-f1.gif) | ||
| Fig. 1 7,7-Disubstituted bicyclo[3.2.0]hept-2-en-6-ones. | ||
In order to investigate the potential of Cunninghamella echinulata 3655 for the resolution of the bicyclo[3.2.0] ketones, resting cells of the fungus were first incubated with three bicyclo[3.2.0]hept-2-en-6-one derivatives; the unsubstituted parent compound 1 (Fig. 1); the 7,7-dimethyl derivative 2 and the 7-endo-phenyl-7-exo-methyl derivative 3, each at a concentration of 0.5 g L−1. Biotransformations of 1 and 3 were continued until the residual ketone constituted approximately 50% of the reaction mixture, in an effort to achieve resolution of the racemic starting materials.
 | ||
| Fig. 2 Baeyer–Villiger, allylic oxidation and reduction products from the biotransformation studies. | ||
The contrast in product profiles as determined by GC for substrates 1, 2 and 3 after 22 h, 48 h and 22 h is illustrated in Fig. 3. Compound 1 was transformed to two major metabolites on GC, being approximately equal amounts of the reduction product 6-endo-hydroxybicyclo[3.2.0]hept-2-ene 4 and a mixture of lactone products, not resolved by GC, but which 1H NMR confirmed to be a 1 : 1.2 mixture of both 2-oxa- 5 and 3-oxa- 6 lactones resulting from non-regioselective oxygen insertion (Fig. 2). These results contrast with those previously reported for biotransformation of 1 with the same organism under equivalent conditions, where only the lactone 5 was obtained.6 The residual ketone 1 was shown to be racemic by chiral GC analysis. 7,7-Dimethylbicyclo[3.2.0]hept-2-en-6-one 2 proved to be a poor substrate for any transformation, but after 48 h incubation, a GC trace showed four products, but of insufficient abundance for characterisation by NMR spectroscopy.
![Contrasting product profiles of biotransformation of bicyclo[3.2.0]hept-2-en-6-one derivatives by Cunninghamella echinulata NRRL 3655: a) GC chromatogram of biotransformation mixture of 1 after 22 h; b) GC chromatogram of biotransformation mixture of 2 after 48 h; c) GC chromatogram of biotransformation mixture of 3 after 22 h.](/image/article/2004/OB/b406016d/b406016d-f3.gif) | ||
| Fig. 3 Contrasting product profiles of biotransformation of bicyclo[3.2.0]hept-2-en-6-one derivatives by Cunninghamella echinulata NRRL 3655: a) GC chromatogram of biotransformation mixture of 1 after 22 h; b) GC chromatogram of biotransformation mixture of 2 after 48 h; c) GC chromatogram of biotransformation mixture of 3 after 22 h. | ||
When C. echinulata was challenged with 7-endo-phenyl-7-exo-methylbicyclo[3.2.0]hept-2-en-6-one 3, one minor and one major product were observed after 22 h biotransformation. NMR spectoscopy confirmed the formation of epoxide 7 (we further prepared this compound independently by treatment of 3 with mCPBA in CH2Cl2, see experimental) and the hydroxylactone 8 (Fig. 2), which was isolated in 41% yield. The latter product represents a hitherto undescribed microbial transformation of the bicyclo[3.2.0]hept-2-en-6-one nucleus and would allow access to a highly elaborated cyclopentanoid ring containing three chiral centres. Hydroxylation in the 2-position yields a hydroxylactone of a substitution pattern previously shown to be of use in the synthesis of, for example, cyclosarkomycin.6 The residual ketone was obtained in 40% yield, but chiral GC analysis showed it to be racemic.
The hydroxylactone 8 is evidently the result of both Baeyer–Villiger oxidation and allylic hydroxylation, accompanied by transposition of the double bond. We postulated that 8 may have arisen first from cytochrome P450 dependent epoxidation of the double bond of 3, followed by base catalysed elimination on the resulting identified epoxide 7 followed by oxygenation (Fig. 4).
 | ||
| Fig. 4 Possible route to the hydroxylactone 8. | ||
This hypothesis appeared to be supported by the inhibition of biotransformation activity in the presence of 1 mM 1-aminobenzatriazole, a cytochrome P450 inhibitor. However, when 7 was isolated, and resubmitted to active fungus under the same reaction conditions, no biotransformation was observed, suggesting that 7 is not an intermediate to 8. The active fungus was then challenged with the 3-oxa lactone derivative 9. As abiotic methods proved unsuccessful at producing 9 from 3, it was produced by a biocatalytic process which will be described elsewhere. Lactone 9 was not transformed to 8 using a fungal culture whose capacity for the transformation of 3 to 8 was monitored in parallel as a control. It is difficult at this stage to suggest whether the lactone 9 is produced first, thence hydroxylated, or allylic hydroxylation to give 15, followed by Baeyer–Villiger oxidation occurs, as there were no other observable abundant intermediates by GC analysis during accumulation of product 8. Subjecting 7-phenyl-7-chlorobicyclo[3.2.0]hept-2-en-6-one 10 to the assay produced a number of products which were not separable by chromatography.
7,7-Dichlorobicyclo[3.2.0]hept-2-en-6-one 12 was rapidly converted to the racemic endo-alcohol 13, with no oxidation products observed. It is perhaps unsurprising that 7,7-diphenylbicyclo[3.2.0]hept-2-en-6-one 14 was not biotransformed under these conditions. Notwithstanding the poor solubility of this compound under the reaction conditions, it may prove sterically demanding for enzymes catalysing either reduction, Baeyer–Villiger oxidation or indeed allylic hydroxylation.
In conclusion, a biocatalytic method for the one step double oxidation of 7-phenylbicyclo[3.2.0]hept-2-en-6-one derivatives has been described, leading to hydroxylactones which may furnish potentially useful chiral cyclopentanone scaffolds for further synthetic elaboration. In addition to this, the investigation has highlighted a hitherto undescribed biotransformation of the bicyclo[3.2.0]hept-2-en-6-one nucleus, which may involve previously undescribed enzyme activities of potential synthetic utility. In due course the use of 8 as an intemediate to various novel chiral ligands will be reported.
Experimental
Procedure for biotransformation of 3
Cunninghamella echinulata NRRL 3655 was obtained from the US Department of Agriculture Agricultural Research Service Culture Collection, Peoria, Illinois USA. A 250 mL flask charged with 60 mL growth medium (corn steep solids, 7.5 g L−1; glucose 10 g L−1, pH 4.85) was inoculated with 0.5 mL of a suspension derived from a 5 mm × 5 mm square of the fungus grown on potato dextrose agar homogenised in 1 mL of sterile water. After three days growth on an orbital shaker at 28 °C, this culture was used to inoculate a 2 L flask charged with 500 mL growth medium. After a further three days growth at 28 °C, the mycelium was filtered off and washed with 50 mM phosphate buffer pH 8.0. The mycelium was resuspended in 500 mL buffer and to this was added substrate (250 mg, 1.26 mmol) dissolved in ethanol (2 mL). The biotransformation was monitored by GC and after 22 h, the mycelium was removed by filtration and the liquor extracted with diethyl ether (3 × 150 mL). The combined organic phases were dried over anhydrous magnesium sulfate and the solvent removed to leave a residue, which was chromatographed over silica to yield, in order of elution, 3 (40%), 7 (7%) and 8 (41%).(±)-7-endo-Phenyl-7-exo-methylbicyclo[3.2.0]hept-2-en-6-one (3)12
2-Methylphenyl acetyl chloride (3.01 g, 17.9 mmol) was reacted with freshly distilled 1,3-cyclopentadiene (2.6 g, 39.3 mmol, 2.2 equiv.) and Et3N (2.7 g, 26.8 mmol, 1.5 equiv.) in hexane (100 mL) at room temperature. After 6 h the mixture was poured through Celite® and washed with hexane. The hexane was removed in vacuo to afford a dark brown oil. Purification by flash chromatography using 15% diethyl ether/hexane gave the title compound (1.27 g, 36%) as a lime green oil. νmax (neat)/cm−1 3059, 2960, 2922, 2852, 1771 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O), 1602 (CC). δH
(270 MHz, CDCl3) 7.15 (5H, m, Ph), 5.55 (1H, dddd, 3J3H
= 6.5, 3J1H
= 1.8, 4J4Hendo
= 1.8, 4J4Hexo
= 1.8, 2-H), 5.47 (1H, dddd, 3J2H
= 6.5, 3J4Hendo
= 1.8, 3J4Hexo
= 1.8, 4J1H
= 1.8, 3-H), 3.99 (1H, ddd, 3J4Hexo
= 8.06, 3J1H
= 7.2, 3J4Hendo
= 1.1, 5-H), 3.55 (1H, dddd, 3J5H
= 7.2, 4J4Hendo
= 3.1, 4J4Hexo
= 1.8, 3J2H
= 1.8, 1-H), 2.63 (1H, ddddd, 2J4Hexo
= 17.6, 4J1H
= 3.1, 3J3H
= 1.8, 4J2H
= 1.8, 3J5H
= 1.1, 4-H endo), 2.41 (1H, ddddd, 2J4Hendo
= 17.6, 3J5H
= 8.06, 3J3H
= 1.8, 4J2H
= 1.8, 4J1H
= 1.8, 4-H exo), 1.67 (3H, s, C7(C-H3)). δC
(68 MHz, CDCl3) 211.1 (4°, C-6), 140.7 (C-1′
), 133.2, 128.2 (2C), 127.4 (2C), 126.2, 126.1, 71.5 (4°, C-7), 57.2 (C-5), 51.6 (C-1), 34.2 (C-4), 27.1 (C7(CH3)). LRMS (EI)
m/z 198 (M+, 15), 183 (M+
− CH3, 12), 170 (M+
− CO, 10), 132 (Ph(CH3)CCO, 45), 104 (Ph(CH3)C, 100), 77 (Ph, 40), 66 (C5H6, 33), 51 (C4H3, 14).
O), 1602 (CC). δH
(270 MHz, CDCl3) 7.15 (5H, m, Ph), 5.55 (1H, dddd, 3J3H
= 6.5, 3J1H
= 1.8, 4J4Hendo
= 1.8, 4J4Hexo
= 1.8, 2-H), 5.47 (1H, dddd, 3J2H
= 6.5, 3J4Hendo
= 1.8, 3J4Hexo
= 1.8, 4J1H
= 1.8, 3-H), 3.99 (1H, ddd, 3J4Hexo
= 8.06, 3J1H
= 7.2, 3J4Hendo
= 1.1, 5-H), 3.55 (1H, dddd, 3J5H
= 7.2, 4J4Hendo
= 3.1, 4J4Hexo
= 1.8, 3J2H
= 1.8, 1-H), 2.63 (1H, ddddd, 2J4Hexo
= 17.6, 4J1H
= 3.1, 3J3H
= 1.8, 4J2H
= 1.8, 3J5H
= 1.1, 4-H endo), 2.41 (1H, ddddd, 2J4Hendo
= 17.6, 3J5H
= 8.06, 3J3H
= 1.8, 4J2H
= 1.8, 4J1H
= 1.8, 4-H exo), 1.67 (3H, s, C7(C-H3)). δC
(68 MHz, CDCl3) 211.1 (4°, C-6), 140.7 (C-1′
), 133.2, 128.2 (2C), 127.4 (2C), 126.2, 126.1, 71.5 (4°, C-7), 57.2 (C-5), 51.6 (C-1), 34.2 (C-4), 27.1 (C7(CH3)). LRMS (EI)
m/z 198 (M+, 15), 183 (M+
− CH3, 12), 170 (M+
− CO, 10), 132 (Ph(CH3)CCO, 45), 104 (Ph(CH3)C, 100), 77 (Ph, 40), 66 (C5H6, 33), 51 (C4H3, 14).
(±)-2,3-exo-Epoxy-7-endo-7-exo-methyl-phenylbicyclo[3.2.0]hept-6-one (7)13
(−)-6-exo-Hydroxy-4-endo-phenyl-4-exo-methyl-3-oxabicyclo[3.3.0]oct-7-en-2-one (8)
[α]D20 = −12.0 (c 0.02, CH2Cl2).14νmax (CH2Cl2)/cm−1 3591, 2927, 2856, 1772, 1603, 1496, 1006. δH (270 MHz, CDCl3) 7.18 (5H, m, Ph), 5.88 (1H, ddd, 3J8H = 5.6, 3J6H = 3.3, 4J5H = 1.9, 7-H), 5.81 (1H, ddd, 3J1H = 5.9, 3J7H = 5.6, 4J6H = 2.2, 8-H), 4.87 (1H, dd, 3J7H = 3.3, 4J8H = 2.2, 6-H), 3.98 (1H, d, 3J8H = 5.9, 1-H), 3.67 (1H, m, 5-H), 1.68 (3H, s, C4(C-H3)). δC (68 MHz, CDCl3) 180.30 (4°, C-2), 140.71 (4°, Ph), 136.91, 135.75, 128.33 (2C), 126.66, 125.80, 76.53 (C-6), 68.71 (4°, C-4), 67.22 (C-1), 50.43 (C-5), 26.91 (C-4(CH3)). LRMS (CI) m/z 248 (M+ = NH4, 30) 230 (M+, 16), 214 (34), 196 (10), 186 (33), 169 (100), 132 (11), 120 (5). HRMS (CI) calculated for C14H14O3, 230.26588; Found 230.26594.

Acknowledgements
I. J. S. F. and G. G. thank the University of York for funding through the I. R. P. F. We thank the EPSRC for a studentship (S.G.) and Stylacats for CASE funding.Notes and references
- For a recent review see L. R. Lehman and J. D. Stewart, Curr. Org. Chem., 2001, 4, 439–470 Search PubMed.
- A. M. Abel, G. R. Allan, A. J. Carnell and J. A. Davis, Chem. Commun., 2002, 1762 RSC.
- A. R. S. Ibrahim, A. M. Galal, M. S. Ahmed and G. S. Mossa, Chem. Pharm. Bull., 2003, 51, 203 CrossRef.
- G. J. Grogan and H. L. Holland, J. Mol. Catal. B—Enzym., 2000, 9, 1 CrossRef CAS.
- M. D. Mihovilovic, B. Muller and P. Stanetty, Eur. J. Org. Chem., 2002, 22, 3711 CrossRef.
- L. Andrau, J. Lebreton, P. Viazzo, V. Alphand and R. Furstoss, Tetrahedron Lett., 1997, 38, 825 CrossRef CAS.
- E. J. Corey, N. M. Weishenker and T. K. Schaaf and W. Huber, J. Am. Chem. Soc., 1969, 91, 5675 CrossRef CAS; C. B. Chapleo, M. A. Finch, T. V. Lee, S. M. Roberts and R. F. Newton, J. Chem. Soc., Perkin Trans. 1, 1980, 2084 RSC.
- J. Lebreton, V. Alphand and R. Furstoss, Tetrahedron, 1997, 53, 145 CrossRef CAS.
- M. E. B. Smith, M. C. Lloyd, N. Derrien, R. C. Lloyd, S. J. C. Taylor, D. A. Chaplin, G. Casy and R. McCague, Tetrahedron Lett., 2001, 12, 703 CrossRef CAS.
- I. J. S. Fairlamb, S. Grant and S. M. Roberts, 2004, unpublished results.
- V. Alphand, A. Archelas and R. Furstoss, Tetrahedron Lett., 1989, 30, 3663 CrossRef CAS; R. Gagnon, G. Grogan, M. S. Levitt, S. M. Roberts, P. W. H. Wan and A. J. Willetts, J. Chem. Soc., Perkin Trans. 1, 1994, 2537 RSC.
- I. J. S. Fairlamb, J. M. Dickinson, R. O'Connor, S. Higson, L. Grieveson and V. Marin, Bioorg. Med. Chem., 2002, 10, 2641 CrossRef CAS.
- I. J. S. Fairlamb, Ph. D. Thesis, Manchester Metropolitan University(UK), 2000.
- We have so far been unable to determine the optical purity or absolute configuration of compound 8. We are currently identifying a general synthetic route to racemic 8, which will help us to determine this. Furthermore, we hope to derivatise 8 to faciltate a determination of the absolute configuration. These studies are ongoing and will be reported in a full paper.
| This journal is © The Royal Society of Chemistry 2004 |