NMR structure determination and calcium binding effects of lipopeptide antibiotic daptomycin
Lee-Jon
Ball
,
Catherine M.
Goult
,
James A.
Donarski
,
Jason
Micklefield
and
Vasudevan
Ramesh
*
Department of Chemistry, University of Manchester Institute of Science and Technology, PO Box 88, Manchester, UK M60 1QD. E-mail: vasudevan.ramesh@umist.ac.uk
First published on 15th June 2004
Abstract
Daptomycin is an acidic lipopeptide antibiotic, whose three-dimensional structure and mechanism of action is currently unknown. Recently daptomycin, trade name Cubicin, was approved as a drug for the treatment of skin-related infections (M. Larkin Lancet, 2003, 3, 677) and became the first antibiotic of its class to be used in the clinic (A. Raja et al., Nature Rev. Drug Discov., 2003, 2, 943–944). We have carried out a systematic high field NMR study of daptomycin and its binding to calcium ions which is essential for antibiotic activity. In this first report, we demonstrate the sequence-specific resonance assignment of daptomycin under resolved NMR measurement conditions. In addition to this, we have determined the 3D structure of apo-daptomycin and demonstrated a 1 : 1 stoichiometry on the binding to calcium ions. We have also demonstrated that the binding of calcium ions does not result in major conformational changes, but does induce aggregation. This may be an important factor in the mode of action of daptomycin.
Introduction
Daptomycin is an acidic lipopeptide antibiotic produced by Streptomyces roseosporus.1 It possesses potent bactericidal activity against many clinically important gram positive pathogens, including methicillin and vancomycin resistant Staphylococcus aureus strains. Recently, it became the first antibiotic of its class to be approved for clinical use.2,3 A salient feature of daptomycin is its requirement for calcium ions for eliciting biological activity.4 Daptomycin is a cyclic tridecapeptide comprised of several D-configured and non-proteinogenic amino acids, including kynurinine (Kyn), ornithine (Orn), and L-3-methylglutamic acid (MeGlu) (Scheme 1).1 The N-terminus is acylated with a n-decanoyl fatty acid side chain, which is likely to be important in penetrating the cyctoplasmic membrane of gram positive pathogens. Screening of a large number of daptomycin variants with various straight and branched fatty acid side chains identified this moiety as a source of toxicity with the decanoyl group exhibiting the least effects.1 The C-terminal carboxylate (Kyn) is cyclised onto the hydroxyl group of Thr residue at position 4, resulting in a cyclic decapeptide core. In addition to MeGlu there are 3 acidic Asp residues that are presumably important for calcium binding and activity.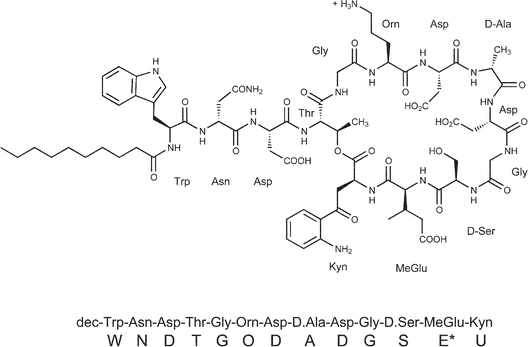 | ||
| Scheme 1 | ||
The mechanism of action and calcium binding sites of daptomycin has not been determined so far. However, two different models for the mode of action have been proposed, which continue to be debated. In the initial model of Boaretti et al.,5 daptomycin inhibits lipoteichoic acid (LTA) biosynthesis in the presence of calcium ions. LTA is a cell surface macromolecule anchored to cytoplasmic membrane, and extends into the peptidoglycan cell wall layer.5 In the second model of Laganas et al.,6 Silverman et al.,7 the long lipid side chain of apo-daptomycin is weakly bound into the cytoplasmic membrane. The binding of calcium ions then causes deeper penetration into the membrane and aggregation of daptomycin which create channels allowing potassium ions to permeate. This leads to membrane depolarisation, which results in disruption of cellular activity and rapid cell death.6,7
Daptomycin shares a similar structure, and possibly a related mode of action, to other acidic lipopeptide antibiotics including the calcium dependent antibiotic CDA,8,9 lipopeptide A-54145,10 friulimicins and amphomycins.11 Noticeably, all of these antibiotics are comprised of a decapeptide lactone or lactam ring derived from cyclisation of L-threonine or L-threo-2,3-diaminobutyrate side chains onto the C-terminal carboxyl group. In addition numerous residues within the cyclic decapeptide cores including acidic residues (Asp and MeGlu) are conserved at the same positions. All of these lipopeptides are biosynthesised on large multi-modular nonribosomal peptide synthetases.12 This mode of biosynthesis means that these peptides are well suited to the biosynthetic engineering methods that lead to new modified peptides.13–16 Indeed the first engineered lipopeptide antibiotics, variants of CDA, were recently generated here at UMIST.9 Understanding the structure–activity relationship of daptomycin and the other acidic lipopeptide is important in the development of new antibiotics by biosynthetic engineering.
In this paper, we provide the results of detailed NMR spectroscopic study of the three dimensional structure of apo-daptomycin in native solution state. The effect on the structure of daptomycin upon binding of calcium ions was monitored by measuring the changes to the pattern of NOEs and to the line widths of assigned protons of several residues.
Results
Daptomycin has a high solubility in water, and hence initial NMR spectra were measured at 5 mM sample concentration, pH 5.0, 298 K (data not shown). However, it was found that the resonance line widths were relatively large for a small peptide of the size of daptomycin signifying the aggregation tendency of the lipopeptide molecule. Accordingly, the sample was diluted to 0.8–1.2 mM, which was found to be optimum in terms of both narrower line widths and spectral sensitivity (data not shown) and this concentration was used for all subsequent NMR experiments. The 1D proton spectrum, in 1H2O, showed resolved resonances with sufficient dispersion for analysis, including a unique low field shifted resonance at 5.48 ppm. This resonance was later assigned (see below) to the side chain Hβ proton of the Thr 4 residue. The low field shift of this proton provides evidence for the ester linkage of Thr residue with aromatic Kyn 13 to form the ten member lactone ring of the cyclic peptide (Scheme 1).Sequence-specific resonance assignment
The identification and sequence-specific assignment of the amino acid spin systems of daptomycin were carried out by a series of 2D experiments according to established methodology.17 For example, the scalar correlated coupled proton connectivities, 3JH–H, which identify the aromatic side chain spin systems of Trp (W1) and Kyn (U13) amino acid residues were analysed using the DQF-COSY18 spectrum measured in 2H2O. Similarly, the selected scalar correlated coupled proton–carbon connectivities, 1JH–C, which identify the long aliphatic side chain spin systems of nonproteinogenic Orn (O6) and MeGlu (E*12) amino acid residues were analysed using the natural abundance, sensitivity-enhanced HSQC19 spectrum measured in 2H2O.The sequence-specific resonance assignment of daptomycin was carried out by measuring its TOCSY20 and NOESY21 spectra in 1H2O (Fig. 1A, B). The TOCSY spectrum (A) shows the intra-residue scalar correlation of exchangeable backbone amide protons with the respective non-exchangeable side chain protons of each residue. Except for the four nearly degenerate amide NH protons at 8.29–8.33, the remaining nine NH protons show resolved unambiguous correlations to the respective vicinal Hα and other long range protons Hβ,Hγ,Hδ of the same spin system. The spectrum also clearly identified the amide protons of the N- and C-terminal residues (Trp and Kyn) as well as the NH proton of the branching Thr residue (Scheme 1), which helped the sequence-specific resonance assignment.
 | ||
| Fig. 1 (A) 600 MHz TOCSY spectrum of daptomycin (1.2 mM) in 90% 1H2O + 10% 2H2O at pH 5.0, 298 K, recorded with a spin-lock mixing time of 80 ms. The spectrum shows the identification of various amino acid spin systems based on scalar correlation of the backbone amide protons of (ω2), with the respective side chain protons (ω1) of each spin system. The connectivities are labelled with the one-letter amino acid code for each residue. The crosspeaks are labelled according to the type of side chain proton suffixed with a Greek letter. (B) 600 MHz NOESY spectrum of daptomycin (1.2 mM) in 1H2O + 10% 2H2O at pH 5.0, 298 K. The spectrum shows sequence-specific resonance assignment by tracing sequential connectivities due to dipolar correlation (NOE) of amide protons (previously identified in the TOCSY spectrum A), with the side chain protons of the neighbouring residue in the sequence. | ||
In the next step, the NOESY spectrum (Fig. 1B) was used to trace the sequence specific resonance assignment of daptomycin. The spectrum shows sequential connectivities due to dipolar correlation (NOE) of amide protons previously identified in the TOCSY spectrum (Fig. 1A), with the side chain protons of the neighbouring residue in the sequence. For example, the resolved low field amide proton of Kyn at 8.52 ppm gives an NOE cross peak to the resolved methyl protons of MeGlu at 0.93 ppm, which resulted in the unambiguous sequence-specific assignment. The 1H and 13C chemical shifts of the assigned resonances of daptomycin are given in Table 1.
| Residue | HN–amide | CHα | CHβ | CHγ | CHδ | Aromatics |
|---|---|---|---|---|---|---|
| Trp 1 | 8.012 | 4.609 | 3.166, 3.255, 29.633 | — | — | 7.258, 7.620, 7.163, 7.438, 7.067, 10.15 (indole HN) |
| Asn 2 | 8.257 | 4.513 | 2.432, 2.519 | — | — | — |
| Asp 3 | 8.291 | 4.686 | 2.665, 2.801 | — | — | — |
| Thr 4 | 8.122 | 4.672 | 5.475, 73.824 | 1.166, 31.087 | — | — |
| Gly 5 | 8.313 | 4.070, 4.018 | — | — | — | — |
| Orn 6 | 8.297 | 4.362, 56.068 | 1.891, 1.793, 30.744 | 1.713 (2), 25.748 | 2.997 (2), 41.693 | — |
| Asp 7 | 8.341 | 4.537 | 2.404, 2.581 | — | — | — |
| D-Ala 8 | 8.187 | 4.318, 52.579 | 1.363 (3), 27.665 | — | — | — |
| Asp 9 | 8.459 | 4.654 | 2.708, 2.774 | — | — | — |
| Gly 10 | 8.338 | 3.992 (2) | — | — | — | — |
| D-Ser 11 | 8.046 | 4.584 | 3.878 (2), 64.188 | — | — | — |
| MeGlu 12 | 8.275 | 4.575 | 2.425, 43.141 | Me 0.931 & 17.147, H2 2.114, 2.369 & 35.918 | — | — |
| Kyn 13 | 8.520 | 4.848, 51.475 | 3.258, 3.535 | — | — | 7.357, 7.412, 6.683, 6.793 |
Structure of apo-daptomycin
The three dimensional structure of apo-daptomycin was determined based on the sequence specific resonance assignment (Table 1) and 142 distance constraints generated from NOESY spectra measured in 1H2O and 2H2O, both with identical mixing time of 200 ms. Based on these distance constraints, a total of 30 structures were calculated, out of which 20 structures with the lowest energy target function were found to be most valid, based on best fitting the allowed angle and upper peak limit files. These 20 were selected for further conformational analysis. The backbone torsion angles (ϕ, Ψ) of the chosen structures were found to be within the steric repulsion limits imposed by a Ramachandran plot22 (data not shown). A total of 38 NOE violations across all the 20 structures were found, the majority being in the structures with the largest energy function. The best quality structure with the lowest energy function, and containing one NOE violation, is illustrated (Figs. 2 and 3). Inspection of the structure shows that apo-daptomycin adopts an extended conformation in solution with turns observed at Ala8 and Gly10/Ser11. Most of the side chain groups protrude out of the ten-member ring and appear exposed to the solvent, whereas the backbone amide groups point inside the ring. The decanoyl chain attached to the amino terminus of Trp 1 is flexible with a high degree of conformational freedom.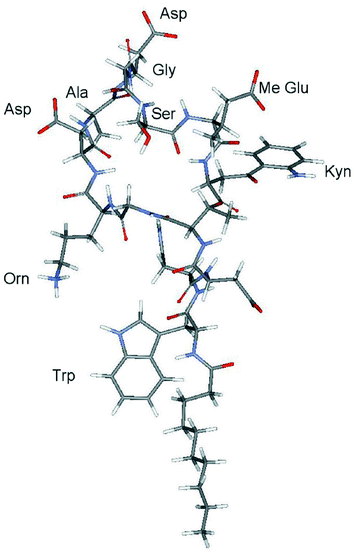 | ||
| Fig. 2 Three-dimensional NMR solution structure of daptomycin with lowest energy target function generated using the NMR structure determination software CYANA. The structure is based on the sequence-specific resonance assignment (Fig. 1) and distance constraints (Table 2) derived from the NOESY spectra of daptomycin (0.8 mM) measured in 1H2O and 2H2O at pH 5.0, 298 K, with identical mixing times of 200 ms. The structure shows an extended conformation for daptomycin with the side chain groups of Asp 7, Asp 9 and MeGlu 12 protruding out of the ring formed by the ester linkage of Kyn 13 with Thr 4. Other structural details are given in the text. | ||
 | ||
| Fig. 3 Distribution of charge (colour coded) on the surface of the structure of apo-daptomycin. Red region denotes negative charge, blue region as positive and white region as neutral. | ||
A histogram correlating the number of constraints arising from each residue of daptomycin is shown in Fig. 4. The data clearly show that the number of long range distance constraints (shaded dark) is very small and that most of the constraints are generated by residues at and adjacent to the terminal residues, and the branching residue Thr 4. The CYANA23 generated distance constraints (number and type) from each spectrum are listed in Table 2.
 | ||
| Fig. 4 A histogram showing the number and range of distance constraints derived from each amino acid residue of daptomycin that were used during the structure determination. The residue labelled C10 refers to the n-decanoyl fatty acid chain attached to the N-terminal Trp 1 (W) residue. The shaded bands correspond to the range of NOE distance constraints: white (intra-residue, n = 0), light grey (sequential, n = 1), dark grey (medium, n > 2) and black (long range, n > 5), where n refers to the number of residues separating the protons giving rise to the distance constraints. | ||
| Spectrum | Type | Distance Calibration Function | Calibration Coefficient (A/B/C) | Average upper-limit distance/Å | Number of Constraints in NOESY |
|---|---|---|---|---|---|
| 1H2O | Backbone | A/r6 | 4.71 × 1011 | 3.60 | 43 |
| 1H2O | Side-chain | B/r4 | 8.17 × 1010 | 4.18 | 16 |
| 1H2O | Methyl | C/r4 | 2.72 × 1010 | 5.53 | 11 |
| 2H2O | Backbone | A/r6 | 1.36 × 1011 | 3.56 | 12 |
| 2H2O | Side-chain | B/r4 | 2.36 × 1010 | 4.76 | 45 |
| 2H2O | Methyl | C/r4 | 7.87 × 109 | 5.32 | 15 |
Effect of calcium binding to daptomycin
The results of the NMR titration of daptomycin with calcium in 2H2O at 293 K are shown in Figs. 5A,B. After the addition of 0.3 molar equivalent of Ca2+, the tendency of resonances to broaden was clearly noticeable in both aromatic and aliphatic regions of the spectrum as exemplified by deterioration in resolution and loss of fine structure. With further addition of Ca2+, increased broadening of resonances was induced and at 1 equivalent of Ca2+, unidentifiable resonance overlap occurred rendering the analysis difficult. Addition of excess of Ca2+ (3 and 10 equivalents) did not cause any further changes to the already severely broadened resonances of daptomycin. Inspection of the titration spectra also showed that the addition of Ca2+ did not induce any discernible changes to the chemical shifts of the resonances. Raising the temperature of measurement from 293 K to 313 K tended to narrow the lines suggesting reduced affinity for Ca2+ at increased temperature. Decreasing the temperature back to 293 K restored the previous broad spectrum of the 1 : 1 complex which indicated that the effect of Ca2+ binding was reversible. | ||
| Fig. 5 (A) 600 MHz 1H-NMR spectra (5.0–8.1 ppm) of the titration of daptomycin (0.8 mM) with molar equivalents of Ca2+ in 2H2O at pH 5.0, 293 K. The spectra show Ca2+ induced increase in resonance line widths of the aromatic protons of Trp 1, Kyn 13 and the Hβ proton of Thr 4 (5.48 ppm) due to aggregation of daptomycin and reaching a maximum at 1 molar equivalent of Ca2+. (B) 600 MHz 1H-NMR spectra (0.5–4.5 ppm) of the titration of daptomycin (0.8 mM) with molar equivalents of Ca2+ in 2H2O at pH 5.0, 293 K. The spectra show Ca2+ induced increase in resonance line widths of the aliphatic protons due to aggregation of daptomycin and reaching a maximum at 1 molar equivalent of Ca2+. | ||
To help monitor changes to the structure of daptomycin upon Ca2+ binding, NOESY spectra of the complex were measured at 293 K. To facilitate data analysis and interpretation, the spectra of daptomycin were measured after the addition of sub-stoichiometric 0.2 and 0.5 molar equivalents of Ca2+ (data not shown), conditions under which the resonance line widths were not excessively broad (Fig. 5A/B). The results showed that, overall, the pattern of NOEs observed was very similar to that of apo-daptomycin and no new NOEs could be identified in the spectra of the complex. This suggested that the structure of daptomycin did not undergo any global conformational change after binding Ca2+. Analogous broadening of labile amide proton resonances was observed in the Ca2+ titration of daptomycin carried out in 1H2O (data not shown).
To correlate the observed calcium induced line width changes noted above to the structure of the antibiotic, the results of the titration are shown by plotting calculated line widths at half height, ν1/2, in Hz (y-axis) versus the molar ratio of Ca2+ ions to daptomycin (x-axis) (Fig. 6). To assist interpretation, only the resolved resonances of the H2 and H4 protons of Trp 1, H3 and H5 protons of Kyn 13 and Hβ proton of Thr 4 were taken up for line shape analysis. As can be seen from the graph, the proton resonances of Trp, Kyn and Thr undergo similar increase in line width after each addition of Ca2+, reaching a plateau at 1 equivalent Ca2+. This indicates that the stoichiometry of Ca2+ binding to daptomycin is 1-to-1.
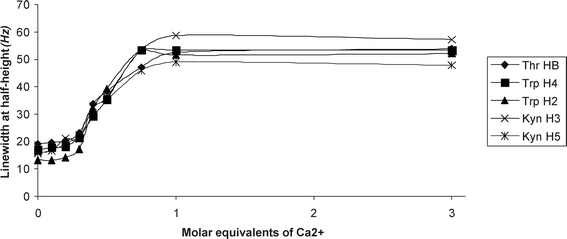 | ||
| Fig. 6 Graph plot showing the increase in resonance line width at half-height (ν1/2, Hz) (y-axis) of daptomycin against the molar equivalents of Ca2+ added (x-axis). The plotted data are derived from Ca2+ titration spectra shown in Fig. 5A. | ||
NMR pH titration of apo-daptomycin
As daptomycin has several acidic residues, which have ionisable side chains with affinity for Ca2+, efforts were made to determine their pKa's by carrying out a one dimensional NMR pH titration in 2H2O over the range of pH 8–pH 2 (Fig. 7). Slight changes to the chemical shifts of resonances could be identified on decreasing the pH from 8 to 4. Further decreasing (up to pH 2) resulted in pronounced broadening of resonances, making the spectral analysis difficult. Presumably protonation of anionic side chain groups reduces the polarity of the peptide, and increases its propensity for intermolecular aggregation which is the cause of this line broadening effect.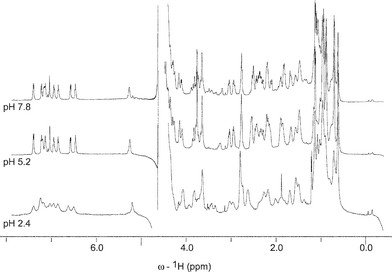 | ||
| Fig. 7 600 MHz 1H-NMR spectra of daptomycin (0.8 mM) in 2H2O, measured at selected pH values 7.8, 5.2 and 2.4, at 293 K. The spectrum at pH 2.4 shows considerable line broadening due to aggregation of daptomycin. | ||
Discussion
The propensity of daptomycin for intermolecular aggregation is readily observed in its NMR spectrum even in the absence of Ca2+. At standard sample concentrations (3–5 mM), best for NMR measurement, apo-daptomycin spectra exhibit broad resonances characteristic of aggregation. The NMR structure determination of daptomycin was aided by careful optimisation of the solution conditions to minimise aggregation, allowing faster tumbling, resulting in narrower line widths of resonances. It was necessary to work at a lower concentration ∼1 mM (pH 5.0, 298 K) to obtain interpretable 2D scalar (TOCSY) and dipolar correlated (NOESY) spectra required for full sequence-specific resonance assignment, and to generate reliable distance constraints from integration of NOE cross peaks. Except for the unusual shifted Hβ resonance (5.45 ppm) of Thr 4, which served as a useful marker, most of the resonances exhibited characteristic chemical shift patterns that one would expect for a peptide of this composition. To enhance the quality of NOE data, a few NOESY spectra were also measured using a newly developed cryoprobe that offered greater sensitivity under the chosen experimental conditions.As noted above, a salient feature of daptomycin is its dependence on Ca2+ for its antibiotic activity. The changes to the NMR resonance line widths of daptomycin on Ca2+ binding provide unambiguous evidence for the affinity between the two. The titration data also showed that after the addition of one molar equivalent of Ca2+, there is no further increase to the resonance line widths. The large resonance line widths of the daptomycin–Ca2+ complex demonstrate that the molecular size of the complex is beyond the simple monomeric form of the complex. In other words, daptomycin adopts a multimeric structure of a defined as yet undetermined size, mediated by an equivalent amount of Ca2+. Except for the changes to line widths, daptomycin does not undergo Ca2+ induced detectable change in chemical shifts or pattern of cross peaks in the NOESY spectra measured with lower ratios of Ca2+. This suggests that the conformation of daptomycin is little affected by binding Ca2+.
The NMR pH titration of apo-daptomycin showed only minor chemical shift changes over the pH range of 2–8, which indicated that the structure of apo-daptomycin determined at pH 5.0 should be similar to the structure at physiological pH (=7.4). Further, the effect of Ca2+ binding on the proton NMR spectra of apo-daptomycin at pH 5 and 7 also were found to be similar, with no detectable changes to the pattern of inter-residue NOEs. These results justify the structure determination of apo-daptomycin at pH 5.0, and enabled us to conclude that this 3D structure is relevant in the mechanism of action of the antibiotic.
The structure of apo-daptomycin is well defined, with a low energy structure that clearly exhibits an extended conformation in solution. The smaller number of long range distance constraints correlate well with the side chain protons protruding out of the ring and exposed to the solvent. Due to severe line broadening effects, resulting in low quality NOESY spectra, it was not possible to determine the corresponding NMR structure of the daptomycin–Ca2+ complex to afford a comparison with the structure of the apo-form. Inspection of the structure of apo-daptomycin shows that the four acidic residues, Asp 3, Asp 7, Asp 9 and MeGlu 12, are not spatially close enough to one another to render a specific binding site for Ca2+, in the absence of a major conformational change. This suggests that the interaction between daptomycin and Ca2+ may simply be electrostatic in nature, aiding aggregation in the process. The decrease in line widths upon raising the temperature (293 K to 313 K) implies a weaker Ca2+ affinity accompanied by breakdown of aggregation.
In summary, we have determined the first 3-D structure of apo-daptomycin and have shown that Ca2+ binds with a 1 : 1 stoichiometry. We have also demonstrated that calcium binding induces aggregation of daptomycin, but does not noticeably alter the conformation of the peptide. The acidic residues (Asp and MeGlu) in the apo-structure are not in close proximity, which suggests that daptomycin is not preorganised for chelation of Ca2+. This suggests that binding to Ca2+ may be weak and electrostatic in nature serving as a neutralising bridge between daptomycin molecules during assembly into a larger complex. The predisposition of daptomycin to aggregate in the presence of calcium supports the membrane depolarisation mode of action.6,7 To fully validate this model, we are currently examining the structure of daptomycin in the presence of model membrane structures as well as with Ca2+.
Experimental
Materials
Daptomycin was kindly supplied by Eli Lilly. The peptide was used without any further purification for NMR investigation. The NMR samples, each using 0.75 mM as a 0.5 ml solution, were prepared by dissolving the antibiotic in either 90% 1H2O + 10% 2H2O solvent mixture or 100% 2H2O. The pH of the sample was adjusted to the desired value using microlitre volumes of concentrated NaOH or HCl. Before doing measurements in 100% 2H2O, the residual 1H2O and labile exchangeable protons were removed by lyophilisation and this was repeated 2–3 times to gain in sensitivity of non-exchangeable aromatic and aliphatic proton resonances.NMR spectroscopy
1D and 2D NMR experiments (DQF-COSY,18 TOCSY,20 NOESY,21 HSQC19) were carried out using a Bruker Avance 600 MHz instrument, equipped with a triple resonance inverse geometry gradient probe and electronic variable temperature unit. Several experiments were also carried out on the recently developed cryoprobe which afforded superior sensitivity. The spectra were acquired using XWIN NMR software interfaced to the spectrometer and the manufacturer supplied pulse programs without any modification. The spectral width varied between 10 and 11 ppm for experiments in 2H2O and 1H2O, respectively, and the transmitter was positioned on the water signal to minimise any artefacts. The large resonance due to the water protons was suppressed by the WATERGATE pulse sequence.24 Usually, 32–64 scans were averaged for each FID before applying suitable apodisation function and then performing the Fourier transformation. 1H and 13C chemical shifts are reported with respect to dilute concentration of internal dioxane (0.01% v/v) used as an internal reference standard.1H–13C correlated sensitivity-enhanced HSQC19 spectrum of daptomycin was measured at natural abundance at 313 K with the coherence delay set to (1/4J =) 145 Hz.19 Most of the 1D NMR data were initially processed using Bruker XWINNMR 3.5 software running on SGI O2 UNIX. For display and analysis purposes, the 1D data were later reprocessed using MestRe-C 3.4 software (http://www.mestrec.com) running on Microsoft Windows XP. Similarly, the 2D NMR data were processed using NMRPipe software (http://spin.niddk.nih.gov/bax/software/NMRPipe)25 running on the RedHat Linux 9.0 operating system (http://www.redhat.com). Usually, each time domain (t1,t2) data was zero-filled twice before apodisation by a shifted sine bell function or a power-sine function, and apodisation before Fourier transformation to yield frequency domain (ω1,ω2) spectra. The processed NMR spectra were displayed, analysed and annotated using SPARKY software (http://www.cgl.ucsf.edu/home/sparky)26 running on Windows XP and Redhat Linux 9.0.
The changes in resonance line widths during the titration of daptomycin with Ca2+ were analysed by fitting them to a Lorentzian curve, using the line shape analysis algorithm available within Mestre-C. After measurement, the algorithm generated output parameters for each fitted peak listing an estimate of its position (chemical shift), intensity, line width at half height (Hz) and the line shape function applied. The fitting procedure however became difficult when the peaks became indistinguishable due to poor signal/noise during later stages of the titration and some of them were adjusted manually to individual line shapes with inevitable errors (5–10%).
Structure calculation
The NMR structure determination of daptomycin was carried out using CYANA 4.0 software23 kindly provided by Dr Peter Guntert under a license agreement. The display and analysis of calculated structures was performed using the VMD 1.8.2 molecular visualisation software27 available from NIH. Modelling of modified amino acid topologies was done using the Hyperchem 7.0 software.The atomic coordinates of the modified amino acids Orn, Kyn, 3-L-MeGlu (3-R and 3-S forms), Thr (esterified to C-terminus), D-Ala and D-Ser were separately built using Hyperchem in PDB format. The topology files carrying the coordinates of the individual amino acid atoms were then converted into CYANA file format. Each modified amino acid topology file (X) was then tested within a Gly-X-Gly tripeptide sequence for any geometrical errors using CYANA's simulated annealing protocol and by introducing fictitious structural constraints. After checking, the topology files were added to the default CYANA library.
The SPARKY assigned NOESY spectra (measured in 1H2O and 2H2O with 200 ms mixing time) were converted into CYANA format. The NOE crosspeak volumes were integrated using gaussian line shape fitting and the data were saved to a peak file. Both the resonance assignment and peak files were converted into CYANA format. Backbone amide NOE distances were generated using A/r6 function and side chain NOE derived distances were generated using B/r4 function. The NOESY spectra in 1H2O and 2H2O produced 70 and 72 distance constraints, respectively. Out of the total 142 constraints, 48 were duplicate and hence discarded. A new temporary distance constraints file was created.
Based on the preliminary distance constraints, a grid search of all possible HABAS angles for backbone and side chains was executed. It was found that one aromatic NOE violated all possible angles and diastereotopic assignments of Trp 1 and so was discarded. The conforming angles from the search were written as a file and the upper bound distance file was modified to take into account the found angles. Based on the distances and angles, and using a random seed of 32568, 30 structures were calculated from which 20 most valid structures were kept. The structure with lowest target function is presented for display and analysis.
Acknowledgments
We thank the EPSRC (U.K.) for the postgraduate studentship to L-JB. We thank Mr R. Banks for helpful discussion on structure determination. We thank Dr A. Golonov, Biomolecular NMR Centre, UMIST for his help with the NMR experiments. We thank Prof. L.Y. Lian for the allocation of NMR time on the 600 MHz NMR spectrometer.References
- M. Debono, B. J. Abbott, R. M. Molloy, D. S. Fukuda, A. H. Hunt, V. M. Daupert, F. T. Counter, J. L. Ott and C. B. Carrell, J. Antiobiot. (Tokyo), 1998, 41, 1093–1105 Search PubMed.
- M. Larkin, Lancet, 2003, 3, 677 CrossRef.
- A. Raja, J. LaBonte, J. Lebbos and P. Kirkpatrick, Nature Rev. Drug. Discov., 2003, 2, 943–944 Search PubMed.
- A. L. Barry, P. C. Fuchs and S. Brown, Antimicrob Agents Chemother., 2001, 45, 1919–1922 CrossRef CAS.
- M. Boaretti, P. Canepari, M. del Mar Lleò and G. Satta, J. Antimicrob. Chemother., 1993, 31, 227–235 CrossRef CAS.
- V. Laganas, J. Alder and J. A. Silverman, Antimicrob. Agents Chemother., 2003, 47, 2682–2684 CrossRef CAS.
- J. A. Silverman, N. G. Perlmutter and H. M. Shapiro, Antimicrob. Agents Chemother., 2003, 47, 2538–2544 CrossRef CAS.
- D. A. Hopwood and H. M. Wright, J. Gen. Microbiol., 1983, 129, 3575–3579 Search PubMed.
- Z. Hojati, C. Milne, B. Harvey, L. Gordon, M. Borg, F. Flett, B. Wilkinson, P. J. Sidebottom, B. A. M. Rudd, M. A. Hayes, C. P. Smith and J. Micklefield, Chem. Biol., 2002, 9, 1175–1187 CrossRef CAS.
- L. D. Boeck, J. Antibiot. (Tokyo), 1990, 43, 587 CAS.
- L. J. Vértesy, E. Ehlers, H. Kogler, M. Kurz, J. Meiwes, G. Seibert, M. Vogel and P. Hammann, J. Antibiot. (Tokyo), 2000, 53, 816–827 CAS.
- M. A. Marahiel, T. Stachelhaus and H. D. Mootz, Chem. Rev., 1997, 97, 2651–2673 CrossRef CAS.
- D. Scwazer, R. Finking and M. A. Marahiel, Nat. Prod. Rep, 2003, 20, 275–287 RSC.
- H. D. Mootz, D. Schwarzer and M. A. Marahiel, ChemBioChem, 2002, 3, 490–504 CrossRef CAS.
- C. T. Walsh, ChemBioChem, 2002, 3, 124–134 CrossRef CAS.
- D. E. Cane, C. T. Walsh and C. Khosla, Science, 1998, 282, 63–68 CrossRef CAS.
- K. Wuthrich, NMR of Proteins and Nucleic Acids, John Wiley & Sons, 1986 Search PubMed.
- M. Rance, O. W. Sorensen, G. Bodenhausen, G. Wagner, R. R. Ernst and K. Wuthrich, Biochem. Biophys. Res. Commun., 1983, 117, 479–485 CrossRef CAS.
- S. Schleucher, M. Schwendinger, M. Sattler, P. Schmidt, O. Schedletzky, S. J. Glaser, O. W. Sorensen and C. Griesinger, J. Biomol. NMR, 1994, 4, 301–306 CrossRef CAS.
- A. Bax and D. G. Davis, J. Magn. Reson., 1985, 65, 355–360 CAS.
- G. Bodenhausen, H. Kogler and R. R. Ernst, J. Magn. Reson., 1984, 58, 370–388 CAS.
- G. N. Ramachandran, C. Ramakrishnan and V. Sasisekharan, J. Mol. Biol., 1963, 7, 95.
- T. Hermann, P. Guntert and K. Wuthrich, J. Mol. Biol., 2002, 319, 209–227 CrossRef.
- M. Piotto, V. Saudek and V. Skelenar, J. Biomol. NMR., 1992, 2, 661–666 CAS.
- F. Delaglio, S. Grzsiek, G. W. Vuister, G. Zhu, J. Peifer and A. Bax, J. Biomol. NMR, 1995, 6, 277–293 CrossRef CAS.
- T. D. Goddard and D. G. Kneller, SPARKY 3, University of California, San Francisco, 2000.
- W. Humphrey, A. Dalke and K. Schulten, J. Mol. Graphics, 1996, 14, 33–38 CrossRef.
| This journal is © The Royal Society of Chemistry 2004 |