Tandem oxidation processes for the preparation of nitrogen-containing heteroaromatic and heterocyclic compounds
Steven A.
Raw
,
Cecilia D.
Wilfred
and
Richard J. K.
Taylor
*
Department of Chemistry, University of York, Heslington, York, UK YO10 5DD. E-mail: rjkt1@york.ac.uk; Fax: +44(0)1904 434 523; Tel: +44(0)1904 432 606
First published on 11th February 2004
Abstract
α-Hydroxy ketones undergo manganese dioxide-mediated oxidation followed by in situ trapping with aromatic or aliphatic 1,2-diamines to give quinoxalines or dihydropyrazines, respectively, in a one-pot procedure which avoids the need to isolate the highly reactive dicarbonyl intermediates. The scope and limitations of these procedures are outlined and modifications to this procedure are discussed in which reduction is carried out in the same reaction vessel, generating piperazines, or oxidation, leading to pyrazines.
Introduction
Nitrogen-containing heteroaromatic and heterocyclic compounds are indispensable structural units for both the chemist and the biochemist. Quinoxalines constitute the basis of many insecticides, fungicides, herbicides and anthelmintics, as well as being important in human health and as receptor antagonists.1,2 There are numerous methods of preparing quinoxalines but the double condensation of a 1,2-dicarbonyl compound and a 1,2-diaminoaromatic is commonly employed.1–3 Similarly, dihydropyrazines, piperazines and pyrazines are of great importance in natural products and as chemotherapeutic agents2b,4 and can be prepared from the corresponding 1,2-dicarbonyl compound and an aliphatic 1,2-diamine,3a,4,5 followed by reduction or oxidation of the resulting dihydropyrazines, if required.We have recently developed a number of manganese dioxide-mediated tandem oxidation processes (TOPs) for the elaboration of alcohols.6–8 As part of this programme, we established that α-hydroxyketones undergo in situ oxidation-trapping when treated with manganese dioxide in the presence of stabilised Wittig reagents, giving γ-ketocrotonates (Scheme 1a).7 In addition, we have also developed a TOP-amine trapping sequence leading to imines (Scheme 1b).8 We therefore decided to extend these procedures and investigate the conversion of α-hydroxyketones 1 into diamino heterocycles.9
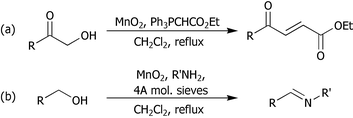 | ||
| Scheme 1 TOP approaches to γ-ketocrotonates and imines. | ||
Results and discussion
We first studied the preparation of quinoxalines 4 and related heterocycles employing a manganese dioxide-mediated TOP with suitable 1,2-diaminoaromatics 2 avoiding the need to isolate the “hyper-reactive”10 1,2-dicarbonyl intermediate 3 (Scheme 2).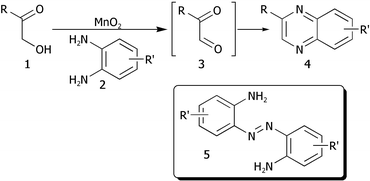 | ||
| Scheme 2 Proposed TOP synthesis of quinoxalines. | ||
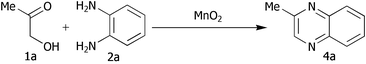
Preliminary studies were carried out using hydroxyacetone 1a
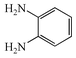
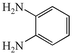
(R![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) Me) and o-phenylenediamine 2a
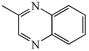
(R′H), and are summarised in Table 1. On the first attempt we were delighted to observe the formation of 2-methylquinoxaline 4a11a but the yield of 43% was disappointing (Table 1, entry i). Addition of acid or base (entries ii and iii), or changing the solvents (entries iv and v) gave no improvement. The major by-product was isolated and identified as the known12 diazobenzene 5a resulting from oxidative coupling of 2a. We therefore investigated batchwise addition of the reagents and the use of different stoichiometries (entries vi and vii). The optimum conditions involved the use of a two fold excess of diamine 2a
(entry vii); in this case the reaction was complete in just one hour and 2-methylquinoxaline 4a was isolated in 79% yield (after chromatography to remove 5a and polymeric by-products). The operational simplicity of this process is noteworthy: after the reaction is complete the remaining oxidant and its by-products are removed by filtration and concentration in vacuo followed by chromatography gives the pure product.
Me) and o-phenylenediamine 2a
(R′H), and are summarised in Table 1. On the first attempt we were delighted to observe the formation of 2-methylquinoxaline 4a11a but the yield of 43% was disappointing (Table 1, entry i). Addition of acid or base (entries ii and iii), or changing the solvents (entries iv and v) gave no improvement. The major by-product was isolated and identified as the known12 diazobenzene 5a resulting from oxidative coupling of 2a. We therefore investigated batchwise addition of the reagents and the use of different stoichiometries (entries vi and vii). The optimum conditions involved the use of a two fold excess of diamine 2a
(entry vii); in this case the reaction was complete in just one hour and 2-methylquinoxaline 4a was isolated in 79% yield (after chromatography to remove 5a and polymeric by-products). The operational simplicity of this process is noteworthy: after the reaction is complete the remaining oxidant and its by-products are removed by filtration and concentration in vacuo followed by chromatography gives the pure product.
| Entry | Reagents | Conditions | Time/h | Yield (%) |
|---|---|---|---|---|
| a Based on 1 eq. alcohol 1a unless otherwise stated; the diazo compound 5a was also formed (up to 15%). b Yield based on diamine 2a. c The isolated yield of diazo compound in this case was 6% based on 2a. | ||||
| i | 15 eq. MnO2, 1.1 eq. 2a | CH2Cl2, Δ | 20 | 43 |
| ii | 15 eq. MnO2, 1.1 eq. 2a, 1% AcOH | CH2Cl2, Δ | 20 | 10 |
| iii | 15 eq. MnO2, 1.1 eq. 2a, 1% K2CO3 | CH2Cl2, Δ | 20 | 10 |
| iv | 15 eq. MnO2, 1.1 eq. 2a | Et2O, Δ | 20 | 12 |
| v | 15 eq. MnO2, 1.1 eq. 2a | THF, Δ | 20 | 15 |
| vi | 15 eq. MnO2, 3.0 eq. 1a, 1.0 eq. 2a | CH2Cl2, Δ | 20 | 51b |
| vii | 10 eq. MnO2, 2.0 eq. 2a, 4 Å mol. sieves | CH2Cl2, Δ | 1 | 79c |
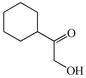
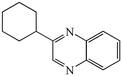
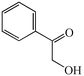
With the successful optimisation results in hand, we moved on to investigate the scope of the process (Table 2), first in terms of α-hydroxyketone substrate (entries i–v). As can be seen, in addition to α-hydroxyacetone 1a, the related 1-cyclohexyl-2-hydroxyethanone 1b13 also gave an excellent yield of the corresponding quinoxaline 4b on treatment with manganese dioxide and 1,2-diaminobenzene. Substrates 1a and 1b were of particular interest as the intermediate α-keto aldehydes are often problematic in terms of the “hyper-reactivity” of the aldehyde function.10 We next moved on to examine aryl substituted α-hydroxyketones (entries iii and iv), both hydroxyacetophenone 1c and the related furyl system 1d13 giving the expected adducts 4c and 4d, respectively. The secondary alcohol benzoin 1e was also shown to react well under these oxidative-trapping conditions with 1,2-diaminobenzene 2a giving 2,3-diphenylquinoxaline 4e in 75% yield (entry v).
| Entry | α-Hydroxy ketone 1 | Amine 2 | Product 4 | Yield (%) |
|---|---|---|---|---|
| a Using 10 eq. MnO2 and 2 eq. diamine in CH2Cl2 at reflux; diazo and polymeric byproducts were removed during chromatography. Yield refers to isolated, chromatographically and spectroscopically pure product. b Isolated as a mixture of regioisomers, ∼7 : 1 in favour of the 3-methylpyrido[2,3-b]pyrazine as determined by 1H NMR spectroscopy c Isolated as a mixture of regioisomers, ∼2 : 1 in favour of the 3-phenylpyrido[2,3-b]pyrazine as determined by 1H NMR spectroscopy. | ||||
| i |
 1a
1a
|
 2a
2a
|
 4a
11a
4a
11a
|
79 |
| ii |
 1b
1b
|
 2a
2a
|
 4b
11b
4b
11b
|
78 |
| iii |
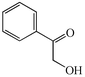 1c
1c
|
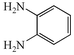 2a
2a
|
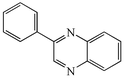 4c
11c
4c
11c
|
79 |
| iv |
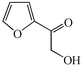 1d
1d
|
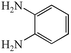 2a
2a
|
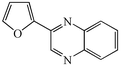 4d
11c
4d
11c
|
89 |
| v |
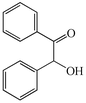 1e
1e
|
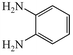 2a
2a
|
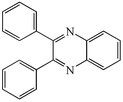 4e
11d
4e
11d
|
75 |
| vi |
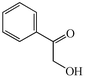 1c
1c
|
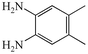 2b
2b
|
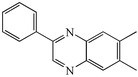 4f
2c
4f
2c
|
66 |
| vii |
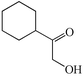 1b
1b
|
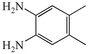 2b
2b
|
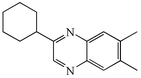 4g
4g
|
89 |
| viii |
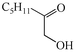 1f
1f
|
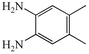 2b
2b
|
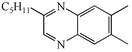 4h
4h
|
62 |
| ix |
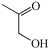 1a
1a
|
 2c
2c
|
 4i
11e
4i
11e
|
66b |
| x |
 1c
1c
|
 2c
2c
|
 4j
2c
11f
4j
2c
11f
|
69c |
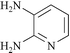
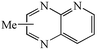
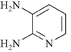
We next studied the use of other diamines (entries vi–x). Thus, 1,2-diamino-4,5-dimethylbenzene 2b was also shown to work well in these reactions giving the desired 2,6,7-trisubstituted quinoxalines 4f, 4g and 4h in good to excellent yields with α-hydroxyketones 1c, 1b and 1f, respectively (entries vi–viii). Finally, 2,3-diaminopyridine 2c was investigated as a coupling partner with 1a and 1c, giving the corresponding 2/3-substituted pyrido[2,3-b]pyrazines 4i and 4j as mixtures of regioisomers (entries ix and x).
Finally in this section of the research, we attempted to apply this methodology to the synthesis of quinoxalin-2-ones. Hence, methyl glycolate (1.20 equivalents) and 1,2-phenylene diamine 2a were reacted under the standard conditions. Unfortunately, oxidation of the glycolate is much slower than for the α-hydroxyketones and therefore, production of diazobenzene is much faster than formation of the quinoxalinone 4k, which was isolated in a disappointing yield of 36% (Scheme 3). Even when the glycolate was used in two-fold excess with respect to phenylene diamine 2a, 1H NMR spectroscopy of the crude product, after complete consumption of 2a, showed the molar ratio of diazobenzene 5a to quinoxaline 4k to be ∼1.1 : 1.
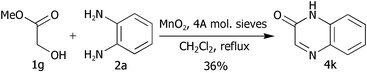 | ||
| Scheme 3 Synthesis of quinoxalin-2-ones. | ||
With a facile and technically straightforward synthesis of quinoxalines and related heteroaromatic compounds from the corresponding α-hydroxyketones to hand, our attention turned to the development of related procedures for the preparation of pyrazines, dihydropyrazines and piperazines.
Initial studies concentrated on the preparation of dihydropyrazines, using α-hydroxyacetophenone 1c as the model α-hydroxyketone with ethylenediamine 5a (Scheme 4). We quickly established that the process was viable under the optimum conditions as used for quinoxaline synthesis. The expected 2-phenyl-5,6-dihydropyrazine 6a was produced in 28% yield without contamination by diazo byproducts. However, somewhat surprisingly, the major product from this reaction was N-benzoyl-N′-formyl 1,2-diaminoethane 7a, obtained in a yield of 50%.
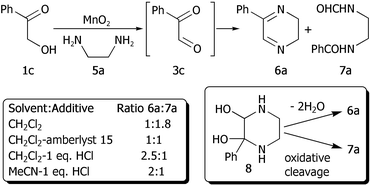 | ||
| Scheme 4 TOP approach to dihydropyrazines. | ||
We assume that a bis-hemi-aminal intermediate, such as 8,5a which would give adduct 6a after dehydration, can also undergo oxidative cleavage in the presence of manganese dioxide giving bis-amide 7a. The oxidative cleavage of 1,2-diols using MnO2 is well known14 but, to our knowledge, it has not been reported with hemi-aminals before. An isolated example of the oxidative cleavage of a bis-hemiaminal using sodium perbromate has however been reported.15 By varying the reaction conditions and solvent (Scheme 4), we found that it was possible to minimise formation of bis-amide 7a by the addition of 2.0 M HCl in Et2O (1 equivalent with respect to the diamine) to the reaction mixture. Using this optimised procedure, dihydropyrazine 6a was obtained in 53% yield. The dihydropyrazines produced in this manner are fairly sensitive, particularly in unpurified form, and were therefore chromatographed immediately after work up using deactivated, neutral alumina.
We then went on to determine the scope of this procedure, particularly in terms of the α-hydroxyketone substrate (Table 3). When using a more hindered diamine, exemplified by (±)-trans-1,2-diaminocyclohexane 5b (entries ii–iv), the dihydropyrazines 6b–6d were obtained in fair to excellent yields, reflecting their greater hydrolytic stability. It should be noted that when using cyclohexanediamine 5b, the bis-amide by-products (corresponding to 9a) were formed in relatively low yields (<15%), and the addition of HCl in Et2O was not required.
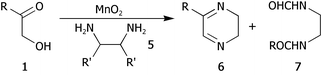
| Entry | R | R′ | Dihydropyrazine | Yield (%) | |
|---|---|---|---|---|---|
| 6 | 7 | ||||
| a Using MnO2 (10 eq.), diamine (1.2 eq.) and powdered 4 Å mol. sieves in CH2Cl2 at reflux; when using diamine 5a, 2.0 eq. of diamine were employed along with 2.0 M HCl in Et2O (1 eq. with respect to diamine). Yield refers to isolated, chromatographically and spectroscopically pure product. b The products were purified immediately after work up (chromatography on deactivated neutral alumina) in order to prevent degradation. | |||||
| i | Phenyl 1c | H5a |
 6a
6a
|
53 | 26 |
| ii | Phenyl 1c | (CH2)45b |
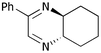 6b
6b
|
64 | 14 |
| iii | 2-Furyl 1d | (CH2)45b |
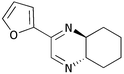 6c
6c
|
64 | 15 |
| iv | Cyclohexyl 1b | (CH2)45b |
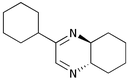 6d
6d
|
53 | 0 |
Having shown that it was possible to produce dihydropyrazines 6a–din situ using TOP methodology, attention moved to the extended one-pot procedures. We recently reported the production of secondary and tertiary amines from activated alcohols using a MnO2 mediated one-pot oxidation imine-formation reduction sequence.8 We envisaged a similar sequence leading from α-hydroxyketones 1 to piperazines 9 (Table 4). Thus, the dihydropyrazine-forming reactions described above were repeated using MnO2–NaBH4. No piperazine formation was observed under these conditions, but the addition of excess methanol to the reaction mixture after dihydropyrazine formation gave the corresponding piperazines 9a–d in good yields (Table 4). As can be seen, the procedure gave good to excellent yields with aromatic (entries i–iii) and aliphatic α-hydroxyketones (entry iv). In the reactions using (±)-trans-1,2-diaminocyclohexane 5b (entries ii–iv), only one diastereomeric product was isolated and we have tentatively assigned these as the all-equatorial adducts shown. There is literature precendence for this selectivity16 and, furthermore, pertinent coupling constants in the 1H NMR spectra were in the region expected for trans-diaxial protons, e.g. for H-2 to H-3axial in compound 9b, the J value is 10.4 Hz.
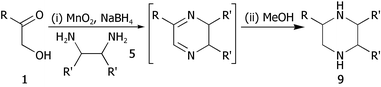
| Entry | R | R′ | Piperazine | Yield (%) |
|---|---|---|---|---|
| a Using MnO2 (10 eq.), diamine (1.2 eq.), NaBH4 (4.0 eq.) and powdered 4 Å mol. sieves in CH2Cl2 at reflux; when using diamine 5a, 2.0 eq. of diamine were employed along with 2.0 M HCl in Et2O (1 eq. with respect to diamine. Yield refers to isolated, spectroscopically pure product. | ||||
| i | Phenyl 1c | H 5a |
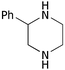 9a
9a
|
52 |
| ii | Phenyl 1c | (CH2)45b |
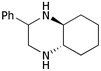 9b
9b
|
75 |
| iii | 2-Furyl 1d | (CH2)45b |
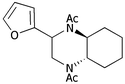 9c
9c
|
60b |
| iv | Cyclohexyl 1b | (CH2)45b |
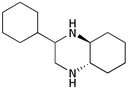 9d
9d
|
84 |
Finally, we investigated the TOP-dihydropyrazine formation-aromatisation sequence leading to pyrazines 10 (Table 5). To this end, the original dihydropyrazine formation was repeated in THF and toluene at reflux for extended periods of time in the presence of excess MnO2 to effect the aromatisation. However, under these conditions, only trace amounts of pyrazines 8 were observed in the toluene reaction. The use of co-oxidants, such as DDQ and CAN, in these reactions resulted in complete degradation of the dihydropyrazines 6. We eventually established that the addition of ∼0.4 M KOH in methanol3a,5b to the refluxing reaction mixture after the formation of dihydropyrazines 6, resulted in production of the corresponding pyrazines 10. It should be noted that the addition of methanol alone did not achieve the desired transformation. The results are summarised in Table 5. As is apparent, the presence of an aromatic substituent facilitates aromatisation (e.g. entries i and iv versus vi and vii).
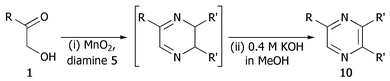
| Entry | R | R′ | Pyrazine | Yield (%) |
|---|---|---|---|---|
| a Using MnO2 (10 eq.), diamine (1.2 eq.) and powdered 4 Å mol. sieves in CH2Cl2 at reflux; when using diamine 5a, 2.0 eq. of diamine were employed along with 2.0 M HCl in Et2O (1 eq. with respect to diamine). After consumption of the hydroxy ketone, KOH in MeOH was added to complete aromatisation. Yield refers to isolated, chromatographically and spectroscopically pure product. | ||||
| i | Phenyl 1c | H 5a |
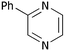 10a
17a
10a
17a
|
45 |
| ii | 4-BrC6H41h | H 5a |
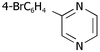 10b
10b
|
57 |
| iii | 2-Furyl 1d | H 5a |
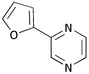 10c
10c
|
60 |
| iv | Phenyl 1c | (CH2)45b |
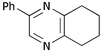 10d
10d
|
66 |
| v | 2-Furyl 1d | (CH2)45b |
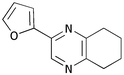 10e
10e
|
64 |
| vi | Cyclohexyl 1b | H 5a |
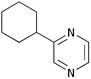 10f
17b
10f
17b
|
33 |
| vii | Cyclohexyl 1b | (CH2)45b |
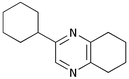 10g
10g
|
0 |
Finally, we examined the value of these methodologies with the complex, multifunctional substrate, hydrocortisone 11. Exposure of 11 to the standard conditions for the formation of quinoxalines, dihydropyrazines, piperazines or pyrazines gave the novel derivatives 12–17 in moderate to excellent unoptimised yields (Scheme 5).
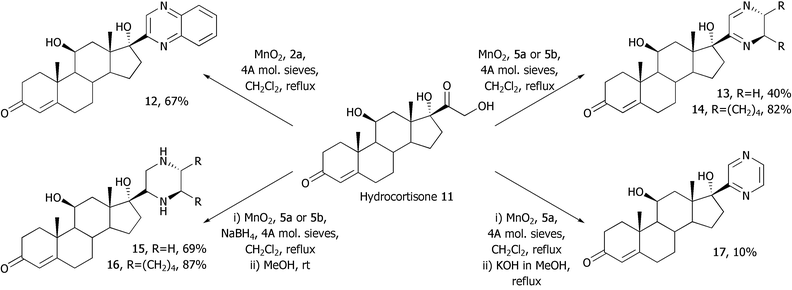 | ||
| Scheme 5 Hydrocortisone as a viable substrate for MnO2-mediated TOP formation of heterocyclic and heteroaromatic functionality. | ||
Conclusion
In conclusion, we have developed a novel methodology for the conversion of α-hydroxy ketones 1 into the corresponding quinoxalines 4 and dihydropyrazines 6via MnO2-mediated tandem oxidation processes with in situ trapping using 1,2-diamines. This methodology has been extended to allow the direct synthesis of piperazines 9 and pyrazines 10 in related TOP sequences from the corresponding α-hydroxy ketones 1 in fair to good yields. These MnO2-TOPs offer the synthetic chemist significant time-cost benefits and, hence, we expect them to find applications in both academic and industrial environments. Further work is continuing to optimise and apply this new chemistry to more complex targets and compound-library synthesis.Experimental
General details: NMR spectra were recorded on Jeol EX-270 and EX-400 instruments using CDCl3 as solvent unless otherwise stated. Tetramethylsilane was used as an internal standard in all cases. IR spectra were recorded on an ATI Mattson Genisis FT-IR or a ThermoNicolet IR100 spectrometer. Low resolution electron impact (EI) mass spectra were recorded on a Kratos MS 25 spectrometer. Chemical ionisation (CI) and high resolution mass spectra were recorded on a Micromass Autospec spectrometer. Flash column chromatography was carried out using Matrex silica gel 60 (70–200) or Brockmann grade 1 neutral alumina, deactivated by the addition of 6 wt% H2O. The α-hydroxy ketones 1b, 1d and 1f were synthesised from the corresponding methyl ketones using the method reported by Moriarty et al.13 All other reagents were purchased from commercial sources and used without further purification. Activated MnO2 was purchased from Aldrich chemical company, catalogue number 21,764–6. PE refers to petroleum ether (boiling point 40–60 °C).Representative procedure for quinoxalines 4
Representative procedure for dihydropyrazines 6
Representative procedure for piperazines 9
Representative procedure for pyrazines 10
11,17-Dihydroxy-10,13-dimethyl-17-quinoxalin-2-yl-1,2,6,7,8,9,10,11,12,13,14,15,16,17-tetradecahydocyclopenta[a]phenanthren-3-one 12
Prepared by the procedure given for 4a using hydrocortisone 1i (0.20 mmol, 0.072 g) and o-phenylenediamine 2a (0.40 mmol, 0.043 g). Purified by column chromatography (EtOAc) to give the title compound12 (0.058 g, 67%) as an orange solid: Rf 0.50 (EtOAc); mp 266–268 °C; νmax (nujol) 3392, 1638, 1276, 1231, 1227, 1112, 942, 865, 775 cm−1; δH (400 MHz) 0.86 (3 H, s, CH3), 1.01 (1 H, dd, J 10.7 Hz, J 3.1 Hz), 1.08–1.18 (1 H, m), 1.31 (1 H, dd, J 14.0 Hz, J 2.1 Hz), 1.37 (3 H, s, CH3), 1.50–1.64 (1 H, m), 1.77 (1 H, dt, J 4.6 Hz, J 13.4 Hz), 1.86–2.51 (12 H, m), 3.04 (1 H, dd, J 12.5 Hz, J 11.9 Hz), 4.41 (1 H, s, OH), 4.61 (1 H, s, OH), 5.61 (1 H, s, CCH), 7.67–7.76 (2 H, m, H-6,7), 7.96–8.07 (2 H, m, H-5,8), 9.01 (1 H, s, H-3); δC
(100 MHz) 18.5, 21.0, 24.3, 31.9, 32.2, 32.8, 33.8, 34.9, 35.0, 39.3, 39.4, 49.1, 51.9, 56.1, 68.4, 84.8, 122.3, 129.0, 129.1, 129.8, 130.3, 140.5, 141.2, 143.9, 157.0, 172.7, 199.8; m/z
(CI) 433 (MH+), 415 (MH+
− H2O)
[HRMS (CI) calcd. for C27H33N2O3 433.2491. Found 433.2497 (1.3 ppm error)].
17-(5,6-Dihydropyrazin-2-yl)-11,17-dihydroxy-10,13-dimethyl-1,2,6,7,8,9,10,11,12,13,14,15,16,17-tetradecahydrocyclopenta[a]phenanthren-3-one 13
Prepared by the procedure given for 6a using hydrocortisone 1i (0.20 mmol, 0.072 g) and ethylenediamine 5a (0.40 mmol, 0.027 mL). Purified by column chromatography (25 : 1 to 15 : 1 CH2Cl2 : MeOH) to give the title compound13 (0.030 g, 40%) as the only isolated product, a pale yellow solid: Rf 0.24 (9 : 1 CH2Cl2 : MeOH); mp 130–132 °C; νmax (nujol) 3404, 1659, 1615, 1590, 1276, 1233, 1189, 1119, 950, 926, 868 cm−1; δH (400 MHz) 0.96 (3 H, s, CH3), 1.01–1.04 (1 H, m), 1.06–1.21 (1 H, m), 1.42 (3 H, s, CH3), 1.34–1.52 (3 H, m), 1.54–1.65 (1 H, m), 1.71–1.95 (3 H, m), 1.96–2.09 (3 H, m), 2.12–2.28 (2 H, m), 2.33 (1 H, m), 2.39–2.53 (2 H, m), 2.73 (1 H, m), 3.34–3.55 (4 H, m, DHP), 3.87 (1 H, br s, OH), 4.43 (1 H, br s, OH), 5.66 (1 H, s, CCH), 8.11 (1 H, d, J 1.5 Hz, imine); δC
(100 MHz) 18.5, 21.1, 24.2, 31.5, 32.2, 32.8, 33.5, 33.9, 35.1, 39.3, 39.9, 43.9, 44.5, 48.5, 51.7, 56.1, 68.5, 84.7, 122.4, 152.6, 162.2, 172.4, 199.7; m/z
(CI) 385 (MH+)
[HRMS (CI) calcd. for C23H33N2O3 385.2491. Found 385.2489 (0.5 ppm error)].
17-(4a,5,6,7,8,8a-Hexahydroquinoxalin-2-yl)-11,17-dihydroxy-10,13-dimethyl-1,2,6,7,8,9,10,11,12,13,14,15,16,17-tetradecahydrocyclopenta[a]phenanthren-3-one 14
Prepared by the procedure given for 6a using hydrocortisone 1i (0.20 mmol, 0.072 g) and trans-1,2-diaminocyclohexane 5b (0.24 mmol, 0.028 mL). Purified by column chromatography (EtOAc to 19 : 1 EtOAc : MeOH) to give the title compound14 (0.072 g, 82%) as an inseparable mixture of diastereomers (∼1 : 1 A : B), a colourless solid: Rf 0.19 (EtOAc); mp 123–125 °C; νmax (nujol) 3393, 1658, 1615, 1584, 1233, 1118, 915, 730 cm−1; δH (400 MHz) 0.88 (3 H, s, CH3A), 0.93 (3 H, s, CH3B), 0.94–1.00 (1 H, m, A + B), 1.02–1.18 (1 H, m, A + B), 1.24–1.57 (6 H, m, A + B), 1.39 (3 H, s, CH3A + B), 1.60–2.06 (11 H, m, A + B), 2.08–2.82 (10 H, m, A + B), 3.68 (1 H, br s, OH, A), 4.39 (1 H, br s, OH, A + B), 4.52 (1 H, br s, OH, B), 5.62 (1 H, s, CCH A + B), 7.98 (1 H, d, J 2.8 Hz, imine A), 8.04 (1 H, d, J 2.7 Hz, imine B); δC
(100 MHz) 18.4/18.5 (CH3), 21.0/21.1 (CH3), 24.0/24.3 (CH2), 25.4/25.5 (CH2), 25.5/25.5 (CH2), 31.4/31.5 (CH), 32.2 (CH2), 32.7/32.8 (CH2), 32.9/33.7 (CH2), 33.0/33.1 (CH2), 33.5/33.5 (CH2), 33.8 (CH2), 34.9 (CH2), 39.2/39.3 (C), 39.5/40.0 (CH2), 48.0/48.9 (C), 51.5/51.6 (CH), 56.1/56.1 (CH), 58.1/58.3 (CH), 58.8/59.1 (CH), 68.2/68.3 (CH), 84.1/84.5 (C), 122.2/122.3 (CH), 151.5/152.4 (CH), 161.7/161.8 (C), 172.6/172.7 (C), 199.7/199.7 (C)
[The pairs of signals were tentatively assigned due to their similar chemical shift and type; however, this is by no means a definitive assignation]; m/z
(CI) 439 (MH+)
[HRMS (CI) calcd. for C27H39N2O3 439.2961. Found 439.2967 (1.4 ppm error)].
11,17-Dihydroxy-10,13-dimethyl-17-piperazin-2-yl-1,2,6,7,8,9,-10,11,12,13,14,15,16,17-tetradecahydocyclopenta[a]phenanthren-3-one 15
Prepared by the procedure given for 9a using hydrocortisone 1g (0.20 mmol, 0.072 g) and ethylenediamine 5a (0.40 mmol, 0.027 mL). Acid/base extraction gave the title compound15 (0.054 g, 69%) as a colourless solid: mp 104–106 °C; νmax (CH2Cl2) 3445, 3053, 2934, 1661, 1451, 1266, 1133, 909, 738, 704 cm−1; δH (400 MHz) 0.73–3.16 (28 H, m), 1.02 (3 H, s, CH3), 1.40 (3 H, s, CH3), 4.33 (1 H, br s, OH), 5.63 (1 H, s, CCH); δC
(100 MHz) 16.9, 21.1, 23.4, 31.3, 32.3, 32.7, 33.9, 35.0, 37.3, 39.3, 41.5, 45.5, 45.8, 46.6, 46.9, 52.1, 56.0, 61.5, 68.2, 83.6, 122.2, 173.0, 199.9; m/z
(CI) 389 (MH+)
[HRMS (CI) calcd. for C23H37N2O3 389.2804. Found 389.2807 (0.8 ppm error)].
17-(Decahydroquinoxalin-2-yl)-11,17-dihydroxy-10,13-dimethyl-1,2,6,7,8,9,10,11,12,13,14,15,16,17-tetradecahydrocyclopenta[a]phenanthren-3-one 16
Prepared by the procedure given for 9a using hydrocortisone 1g (0.20 mmol, 0.072 g) and trans-1,2-diaminocyclohexane 5b (0.24 mmol, 0.028 mL). With trans-1,2-diaminocyclohexane 5b no HCl was used. Acid/base extraction gave the title compound 16 (0.077 g, 89%) as a mixture of several diastereomers (as shown by 1H and 13C NMR spectroscopy), a colourless solid: mp 112–113 °C; νmax (nujol) 3429, 2870, 2727, 1657, 1461, 1377, 749 cm−1; m/z (CI) 443 (MH+) [HRMS (CI) calcd. for C27H43N2O3 443.3274. Found 443.3275 (0.4 ppm error)].11,17-Dihydroxy-10,13-dimethyl-17-pyrazin-2-yl-1,2,6,7,8,9,10,11,12,13,14,15,16,17-tetradecahydocyclopenta[a]phenanthren-3-one 17
Prepared by the procedure given for 10a using hydrocortisone 1g (0.20 mmol, 0.072 g) and ethylenediamine 5a (0.40 mmol, 0.027 mL). Column chromatography (EtOAc to 4 : 1 EtOAc : MeOH) gave the title compound17 (0.008 g, 10%) as a colourless solid: Rf 0.34 (9 : 1 CH2Cl2 : EtOAc); mp 185–190 °C (decomp.); νmax (nujol) 3455, 2953, 1642, 1614, 1234, 1157, 1116, 1915, 859 cm−1; δH (400 MHz) 0.84 (3 H, s, CH3), 1.08 (1 H, m), 1.13–1.34 (3 H, m), 1.43 (3 H, s, CH3), 1.50–1.64 (2 H, m), 1.81–2.12 (6 H, m), 2.12–2.21 (1 H, m), 2.23–2.30 (1 H, m), 2.32–2.40 (1 H, m), 2.42–2.57 (2 H, m), 2.82 (1 H, m), 4.18 (1 H, br s, OH), 4.49 (1 H, br d, J 2.8 Hz, OH), 5.70 (1 H, s, CCH), 8.53 (2 H, br s, H-pyraz.), 8.82 (1 H, s, H-pyraz.); δC
(100 MHz) 18.6, 21.2, 24.3, 32.2, 32.4, 33.0, 34.1, 35.0, 35.2, 39.5, 39.6, 48.7, 51.8, 56.3, 68.8, 84.4, 122.6, 142.6, 143.1, 143.3, 157.2, 172.4, 199.8; m/z
(CI) 383 (MH+)
[HRMS (CI) calcd. for C23H31N2O3 383.2321. Found 383.2331 (2.6 ppm error)].
Acknowledgements
We are grateful to the EPSRC for postdoctoral support (ROPA fellowship, S.A.R.) and to Universiti Teknologi, Petronas, Malaysia for a Ph. D. Scholarship (C.D.W.).References
- G. Sakata, K. Makino and Y. Kurasawa, Heterocycles, 1988, 27, 2481.
- (a) N. Sato, in Comprehensive Heterocyclic Chemistry II, eds. A. R. Katritzky, C. W. Rees, E. F. Scriven, Elsevier Science Ltd., Oxford, 1996vol. 6, ch. 6.03 Search PubMed; for more recent references see; (b) L. E. Seitz, W. J. Suling and R. C. Reynolds, J. Med. Chem., 2002, 45, 5604 CrossRef CAS; (c) A. Gazit, H. App, G. McMahon, J. Chen, A. Levitzki and F. D. Bohmer, J. Med. Chem., 1996, 39, 2170 CrossRef CAS.
- (a) A. Ohto, T. Watanabe, Y. Akita, M. Yoshida, S. Toda, T. Akamatsu, H. Ohno and A. Suzuki, J. Heterocycl. Chem., 1982, 19, 1061 CAS; (b) P. Darkins, N. McCarthy, M. A. McKervey and T. Ye, J. Chem. Soc., Chem. Commun., 1993, 1222 RSC; (c) D. Villemin and B. Martin, Synth. Commun., 1995, 25, 2319 CAS.
- (a) N. Sato, in Comprehensive Heterocyclic Chemistry II, eds. A. R. Katritzky, C. W. Rees, E. F. Scriven, Elsevier Science Ltd., Oxford, 1996,vol. 6, ch. 6.03 Search PubMed; for more recent references see; (b) J. A. Bender, N. A. Meanwell and T. Wang, Tetrahedron, 2002, 58, 3111 CrossRef CAS; (c) C. J. Dinsmore and D. C. Beshore, Tetrahedron, 2002, 58, 3297 CrossRef CAS; (d) R. Gust, R. Keilitz and K. Schmidt, J. Med. Chem., 2002, 45, 2325 CrossRef CAS; (e) A. K. Ghosh, W. J. Thompson, M. K. Holloway, S. P. McKee, T. T. Duong, H. Y. Lee, P. M. Munson, A. M. Smith, J. M. Wai, P. L. Darke, J. A. Zugay, E. A. Emini, W. A. Schleif, J. R. Huff and P. S. Anderson, J. Med. Chem., 1993, 36, 2300 CrossRef CAS; (f) J. W. H. Watthey, T. Gavin, M. Desai, B. M. Finn, R. K. Rodebaugh and S. L. Platt, J. Med. Chem., 1983, 26, 1116 CrossRef CAS; (g) L. J. Street, R. Baker, T. Book, C. O. Kneen, A. M. MacLeod, K. J. Merchant, G. A. Showell, J. Saunders, R. H. Herbert, S. B. Freedman and E. A. Harley, J. Med. Chem., 1990, 33, 2690 CrossRef CAS; (h) F. Navas III, M. J. Bishop, D. T. Garrison, S. J. Hodson, J. D. Speake, E. C. Bigham, D. H. Drewry, D. L. Saussy, J. H. Liacos, P. E. Irving and M. J. Gobel, Bioorg. Med. Chem. Lett., 2002, 12, 575 CrossRef; (i) N. Murugesan, Z. Gu, P. D. Stein, S. Spergel, S. Bisaha, E. C.-K. Liu, R. Zhang, M. L. Webb, S. Moreland and J. C. Barrish, Bioorg. Med. Chem. Lett., 2002, 12, 517 CrossRef CAS.
- (a) L. L. Darko and J. Karliner, J. Org. Chem., 1971, 36, 3810 CrossRef CAS; (b) P. Darkins, M. Groarke, M. A. McKervey, H. M. Moncrieff, N. McCarthy and M. Nieuwenhuyzen, J. Chem. Soc., Perkin Trans. 1, 2000, 381 RSC; (c) T. Yamaguchi, M. Eto, K. Harano, N. Kashige, K. Watanabe and S. Ito, Tetrahedron, 1999, 55, 675 CrossRef CAS; (d) K. Gollnick, S. Koegler and D. Maurer, J. Org. Chem., 1992, 57, 229 CrossRef CAS.
- For oxidation-Wittig trapping see: X. Wei and R. J. K. Taylor, J. Org. Chem., 2000, 65, 616 Search PubMed; L. Blackburn, H. Kanno and R. J. K. Taylor, Tetrahedron Lett., 2003, 44, 115 CrossRef CAS and references therein.
- K. A. Runcie and R. J. K. Taylor, Chem. Commun., 2002, 974 RSC.
- (a) L. Blackburn and R. J. K. Taylor, Org. Lett., 2001, 3, 1637 CrossRef CAS; (b) H. Kanno and R. J. K. Taylor, Tetrahedron Lett., 2002, 43, 7337 CrossRef CAS.
- S. A. Raw, C. D. Wilfred and R. J. K. Taylor, Chem. Commun., 2003, 2286 RSC.
- R. E. Ireland and D. W. Norbeck, J. Org. Chem., 1985, 50, 2198 CrossRef CAS.
- (a) N. L. Maidwell, M. R. Rezai, C. A. Roeschlaub and P. G. Sammes, J. Chem. Soc., Perkin Trans. 1, 2000, 1541 RSC; (b) T. Caronna, A. Citterio, T. Crolla and F. Minisci, J. Chem. Soc., Perkin Trans. 1, 1977, 865 RSC; (c) H. Ihmels, M. Maggini, M. Prato and G. Scorrano, Tetrahedron Lett., 1991, 32, 6215 CrossRef CAS; (d) P. J. Steel and G. B. Caygill, J. Organomet. Chem., 1990, 395, 359 CrossRef CAS; (e) Y. Blache, A. Gueiffier, O. Chavignon, J. C. Teulade, J. C. Milhavet, H. Viols and J. P. Chapat, J. Heterocycl. Chem., 1994, 31, 161 CAS; (f) J. Armand, K. Chekir and J. Pinson, Can. J. Chem., 1978, 56, 1804 CAS.
- I. Bhatnagar and M. V. George, J. Org. Chem., 1968, 33, 2407 CrossRef CAS.
- R. M. Moriarty, B. A. Berglund and R. Penmasta, Tetrahedron Lett., 1992, 33, 6065 CrossRef CAS.
- G. Ohloff and W. Giersch, Angew. Chem., Int. Ed. Engl., 1973, 12, 401 CrossRef; H. S. Outram, S. A. Raw and R. J. K. Taylor, Tetrahedron Lett., 2002, 43, 6185 CrossRef CAS.
- H. Biltz, Justus Liebigs Ann. Chem., 1909, 36, 262.
- (a) M. H. Nantz, D. A. Lee, D. M. Bender and A. H. Roohi, J. Org. Chem., 1992, 57, 6653 CrossRef CAS; (b) L. W. Jenneskens, J. Mahy, E. M. M. de Brabander-van den Berg, I. Van der Hoef and J. Lugtenburg, Rec. Trav. Chim. Pays-Bas, 1995, 114, 97 CAS.
- (a) G. J. Ellames, J. S. Gibson, J. M. Herbert and A. H. McNeill, Tetrahedron, 2001, 57, 9487 CrossRef CAS and references therein; (b) A. Lablache-Combier and B. Planckaert, Bull. Soc. Chim. Fr., 1974, 225.
- A. Fürstner, A. Leitner, M. Mencez and H. Krause, J. Am. Chem. Soc., 2002, 124, 13856 CrossRef.
- P. T. Grovenstein, J. Org. Chem., 1960, 25, 68 CrossRef.
- T. Nishio, J. Org. Chem., 1984, 49, 827 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2004 |