Element selective characterization of stability and reactivity of selenium species in selenized yeast
Peter C.
Uden
*a,
Harriet
Totoe Boakye
a,
Chethaka
Kahakachchi
a,
Rameh
Hafezi
a,
Paula
Nolibos
a,
Eric
Block
b,
Sherida
Johnson
b and
Julian F.
Tyson
a
aDepartment of Chemistry, Lederle Graduate Research Tower A701, University of Massachusetts, Amherst, MA 01003-9336, USA
bDepartment of Chemistry, SUNY-Albany, Albany, NY 12222, USA
First published on 13th October 2003
Abstract
The concerted application of element specific atomic spectral detection for chromatographic eluent monitoring allows previously unexploited qualitative and quantitative analytical concepts to be developed for the determination of selenium species. Selenium speciation is vital in order to better understand its metabolism and biological significance in clinical chemistry, biology, toxicology, and nutrition. Fluoroacid ion pair HPLC with ICP-MS detection and GC derivatization with atomic emission detection (AED) together aid analysis and elucidation of reaction pathways of selenium compounds in high selenium enriched yeast, as used widely in nutritional and clinical cancer preventative studies. Comparisons between currently produced and archived selenized yeasts show major differences in speciation. The formation of selenomethionine selenoxide and the identification of Se–S bonded S-(selenomethyl)-cysteine in archived nutritional yeast may be important for short and long term stability and nutritional activity studies.
Introduction
Selenium has a vital nutritional role and has cancer chemopreventive properties, but the speciation of selenium compounds, which is needed to define their biological roles, presents a great challenge.1 The study of selenium toxicity above the limited range of nutritional necessity also mandates high accuracy and precision in selenium speciation. Ionic selenium and organoselenium compounds, selenoamino acids and related compounds, are present in a diverse range in both enriched and natural matrixes. The human nutritional metabolic need for selenium derives from its incorporation into the active selenol group (–SeH) centers of glutathione peroxidase, thioredoxin reductase and other selenoenzymes.2,3 Cancer chemoprevention is associated with inorganic selenium salts, selenoamino acids and organoselenium compounds such as methaneselenol CH3SeH.4–6 Selenoproteins contain selenium in the form of selenocysteinyl residues; proteins that contain this element in the form of selenomethionyl residues are not formally classified as selenoproteins. Analytical techniques for the determination of selenium species have been reviewed,7–10 but many methods have only been applied to a few commercially available standards. Table 1 shows some selenium species that have attracted nutritional, clinical and bioanalytical attention.| 1 | Selenous acid, selenite | SeO32− | 2 | Selenic acid, selenate | SeO42− |
| 3 | Selenocyanate | SeCN− | 4 | Methaneseleninic acid anion | MeSe(O)O− |
| 5 | Methaneselenenic acid anion | MeSeO− | 6 | Dimethyl selenide | Me2Se |
| 7 | Dimethyl diselenide | Me2Se2 | 8 | Methaneselenol | MeSeH |
| 9 | Trimethylselenonium cation | Me3Se+ | |||
| 10 | Selenocysteine | H3N+–CH(COO−)–CH2–SeH | |||
| 11 | Selenocystine | H3N+–CH(COO−)–CH2–Se–Se–CH2–CH(COO−)–NH3+ | |||
| 12 | Selenomethionine | H3N+–CH(COO−)–CH2–CH2–Se–Me | |||
| 13 | Selenomethionine oxide hydrate | H3N+–CH(COO−)–CH2–CH2–Se(OH)2–Me | |||
| 14 | Se-Methylselenocysteine | H3N+–CH(COO−)–CH2–Se–Me | |||
| 15 | γ-Glutamyl-methylselenocysteine | H3N+–CH2–CH2–CO–NH–CH(COO−)–CH2–Se–Me | |||
| 16 | Selenocystathionine | H3N+–CH(COO−)–CH2–CH2–Se–CH2–CH(COO−)–NH3+ | |||
| 17 | Selenohomocysteine | H3N+–CH(COO−)–CH2–CH2–SeH | |||
| 18 | Se-Adenosylselenohomocysteine | NH2–CH(COOH)CH2CH2SeCH2C4H5O3C5N4NH2 | |||
Treatment of selenium-containing samples with proteolytic enzymes, followed by extraction, enables more than 90% of total selenium to be recovered and speciated, perfluoro acid ion pair reversed phase HPLC giving good separation of selenite, selenate, methaneseleninic acid, selenoamino acids, selenoxides, etc. HPLC interfaced with the inductively coupled plasma mass spectrometer (ICP-MS) gives total element determination and selenium specific detection to low ppb levels and has been applied to a number of natural and enhanced level selenium speciation studies.4,11–17 Volatile compounds such as dialkyl selenides and selenoamino acid derivatives have been determined by headspace–capillary gas chromatography with atomic emission element specific detection (GC-AED).17,18
Selenium-enriched yeast supplements have given statistically significant reductions in cancer incidence and mortality in human intervention trials19 and it has been shown that high-selenium garlic is effective in rat mammary cancer chemoprevention.20 It has been proposed that anti-tumorigenic activity arises from selenomethionine (SeMet) and selenocysteine (SeCys), and methylated forms such as Se-methyl-SeCys that are converted to metabolites, including methaneselenol (CH3SeH).5,21
‘High Selenium Yeast’ was developed to meet the need for a reliable, biologically available source of selenium. Under agreements with the Division of Cancer Prevention, National Cancer Institute (DCP-NCI), clinical trials have been initiated to test hypotheses that Se-supplementation improves health by reducing risks of cancers of the colon, lung and prostate. The Clark study19 is now being extended in the ‘SELECT’-Selenium and Vitamin E Cancer Prevention Trial, which will monitor the incidence of prostate cancer in 32![[hair space]](https://www.rsc.org/images/entities/char_200a.gif) 400 healthy men over a 12-year study period, in which selenomethionine will be employed as the selenium dosage agent.22 The DCP-NCI has noted that, should these studies confirm the previously reported cancer-protective effects of Se, then two further steps will be required in order to ascribe clinical benefit to the dietary supplement. The Se-compounds in the supplement must be identified and evidence of batch-to-batch reproducibility with respect to those Se-compounds must be available. Analyses of commercially available preparations have shown organically-bound Se to vary in range from 0 to 97% of total Se content, with some products showing only a few Se-species and others containing more than 20 different Se-compounds.11,16,23,24
400 healthy men over a 12-year study period, in which selenomethionine will be employed as the selenium dosage agent.22 The DCP-NCI has noted that, should these studies confirm the previously reported cancer-protective effects of Se, then two further steps will be required in order to ascribe clinical benefit to the dietary supplement. The Se-compounds in the supplement must be identified and evidence of batch-to-batch reproducibility with respect to those Se-compounds must be available. Analyses of commercially available preparations have shown organically-bound Se to vary in range from 0 to 97% of total Se content, with some products showing only a few Se-species and others containing more than 20 different Se-compounds.11,16,23,24
A typical commercially available selenized yeast such as SelenoExcell™ (Cypress) has a Se content of 1250 µg g−1 dry matter (±5%), a minimum protein content of 50% (w/w), and a phosphate content in the range of 2.5–3.4% (w/w). However, because it is a biological product, batch-to-batch variation in its composition is possible, requiring informative and rigorous QA/QC protocols. The greater part of the Se in SelenoExcell™ is organically bound, and the major single component (released from the protein-bound form by enzymatic digestion and extraction) is selenomethionine (SeMet), comprising 60–70% of the total Se.11 However, it is possible that selenized yeast may contain organoselenium species or precursors that may be more anti-carcinogenically potent than SeMet.
We now report a more extensive characterization study of a number of yeast samples, including elucidation of changes in Se speciation upon lengthy storage and, in particular, speciation of material used in the human intervention trial of Clark et al.19 The complementary techniques of HPLC-ICP-MS and GC-AED were used to characterize the materials.
Experimental
Instrumentation
An Elan 5000 inductively coupled plasma mass spectrometer (PerkinElmer Sciex, Norwalk, CT, USA) was used for total selenium determination and HPLC-ICP-MS. Samples were introduced using a cross-flow nebulizer and double-pass spray chamber. For total selenium determination 82Se and 77Se were monitored with 74Ge as an internal standard using a 250 ms dwell time (n = 27); isobaric interference from 74Se was automatically corrected as confirmed for certified materials.16 The chromatographic system consisted of a liquid chromatographic pump (SP8810, Spectra-Physics, San Jose, CA, USA), and either a 5 µm Symmetry Shield RP8 bonded hybrid stationary phase column (3.9 mm × 15 cm) or a 5 µm XTerra RP-C18 column ((4.6 mm × 15 cm) Waters Corporation, Milford, MA, USA), that has a polar modifier group between the C8 group and the silica base. The column was connected to the nebulizer with PEEK tubing (30 cm × 0.25 mm id). The mobile phase compositions were as follows: 99 + 1 (v/v) water–methanol was used in each case with (a) 0.1% trifluoroacetic acid (TFA) or (b) 0.1% heptafluorobutanoic acid (HFBA).A Hewlett-Packard HP 5921A atomic emission detector (AED) interfaced with a HP 5890II gas chromatograph was used. The injection port (splitless) was maintained at 250 °C: the GC oven was programmed from 100 °C (initial temperature for 5 min) to 200 °C at 5 °C min−1, holding at 200 °C for 5 min. A HP 1 25 m × 0.32 mm × 0.17 µm (film thickness) column was used. The helium plasma gas flow was kept at 180 ml min−1. Hydrogen was used as reagent gas, with detection at 181 nm (S) and 196 nm (Se).
A HP mass selective detector (Hewlett-Packard Co.) interfaced with a HP 6890 GC was used for GC-MS analysis, using a HP-5 (5% phenyl, 95% polydimethylsiloxane, 30 m × 0.25 mm × 0.25 µm) column. The oven was run at an isothermal temperature of 160 °C and helium was used as a carrier gas at a flow rate of 2 ml min−1. The split ratio was 10 : 1 and the column head pressure was 13.28 psi.
Chemicals
Sodium selenate, sodium selenite, DL-selenomethionine, DL-selenoethionine, DL-selenocystine, Protease XIV, and ethyl chloroformate (ECF) were obtained from Sigma Chemical Company (St. Louis, MO, USA). Se-Methyl-DL-selenocysteine was obtained from Dr. Howard Ganther (University of Wisconsin, Madison, WI, USA). Plasma selenium and germanium standard solutions (1000 µg ml−1) were obtained from Spex Industries Inc., Edison, NJ, USA. Selenium-enriched yeast (SelenoExcell™, also designated SelenoPrecise) was obtained from Cypress Inc., Fresno, CA, USA. Archived selenized yeast and tablets fabricated from it, as employed in the human intervention trials,19 were obtained from the University of Arizona, McKesson Corp. and Cornell University (archive numbered as N1–N13). Stock solutions of selenoamino acids were prepared in 0.2 M HCl. A stock solution of sodium selenate was prepared in 2% (v/v) HNO3, while the plasma selenium standard was used as a stock solution of sodium selenite. Working solutions were diluted with mobile phase and stored in the dark between 0–4 °C. The standards used in measurements were thus 0.02–0.002 M in HCl or 0.0002% in HNO3 after dilution, these acid concentrations having no effect on chromatographic separation and speciation considering the 10 µL volumes injected.Procedure
000 Dalton molecular weight cut-off. Samples were acidified before injection (900 µl of extract plus 100 µl of concentrated HFBA, i.e. 10% HFBA in extract).
000 ppm standard solutions. Then, 1000 ppm derivatized standard solutions were prepared as follows: 100 µl of the 10000 ppm selenoamino acid stock solution was treated with 1 ml of water–ethanol–pyridine mixture (60 ∶ 32 ∶ 8), 50 µl of ECF was added and the mixture shaken until gas evolution ceased. Then, 1 ml of chloroform (containing 1% ECF) was added and the derivatives were extracted into the organic phase which was evaporated to dryness. The residue was redissolved in 1 ml of hexane and 1 µl was injected into the GC-AED or GC-MS.
A 2-ml portion of the aqueous (non-acidified) extract prepared for HPLC analysis was also taken for examination by GC-AED. A water–ethanol–pyridine mixture (60 ∶ 32 ∶ 8 by volume—15 ml) was added followed by ethyl chloroformate (ECF—1 ml), the sample was shaken and any evolved gas vented. Ethylated derivatives were extracted into 3 ml of chloroform (containing 1% ECF) by shaking for 30 min. The chloroform layer was separated, solvent was removed with a flow of nitrogen and the residue was redissolved in 100 µl chloroform or hexane. One µl was injected.
High-performance liquid chromatography provides the core methodology, since some of the target chemical species are not directly amenable to gas phase separations. However, the need is to develop and apply a complementary suite of separation methods which, when applied together, will give the greatest confidence of reliable information.
While ion exchange chromatography has been extensively applied, we have found reversed phase ion pair HPLC to be successful. Incorporation of a fluorinated acid ion-pairing agent to establish a pH of ca. 2–2.5 enables anions such as selenite, selenate, etc., to be protonated completely to give neutral species that are eluted rapidly. Cationic (positive) species, such as trimethylselenonium, are paired with the fluorinated acid anion to give ‘neutral’ ion pairs and are retained somewhat more. Selenoamino acids are protonated and also ‘ion paired’ with the fluoroacid anions and elute at different times depending upon their degree of retention on the stationary phase. Different fluorinated acids provide different degrees of separation in different regions of the chromatogram. Larger molecules, such as peptides and proteins, are not eluted and separated by this procedure, although intermediate species such as Se-adenosyl-selenohomocysteine and γ-glutamyl-Se-methylselenocysteine can be eluted in a reasonable time frame.
Results and discussion
Selenium specific HPLC-ICP-MS chromatograms of enzymatic (Protease XIV) hydrolyzate products from a 1250 ppm reference yeast (SelenoExcell™), obtained on the Symmetry Shield™ column, are shown in Fig. 1. The response scale of the lower chromatogram is expanded approximately 25-fold. In this case selenomethionine is clearly the major selenium species eluted. In terms of percentage selenium distributions, as expressed as total selenium area response of eluted peaks (a valid approach since selenium response is independent of molecular structures), selenomethionine comprised 84% of eluted selenium. The only other positively identified selenium compounds were selenite (0.1%), γ-glutamyl-Se-methyl-selenocysteine (0.5%) and Se-adenosylselenohomocysteine (0.5%), as authenticated by retention of standards and HPLC-MS. We have reported a similar separation for a more highly enriched yeast (1922 ppm Se—Nutrition 21, San Diego, CA).11 For the latter yeast there were indications of other selenium compounds including Se-cystathionine, Se-lanthionine and Se-methylselenocysteine at levels too low for mass spectral confirmation. In these chromatograms an ‘unknown’ selenium peak that does not correspond to any known standards but comprises ca. 5% of eluted selenium, elutes at ca. 2.5 min on the Symmetry Shield™ column (U1). Another small unidentified peak (ca. 2%-eluted selenium) appears at ca.10.5 min (U2). U1 proves to be related to the oxidation of selenomethionine, as is shown subsequently. We show U2 to be a previously unidentified organoselenium species.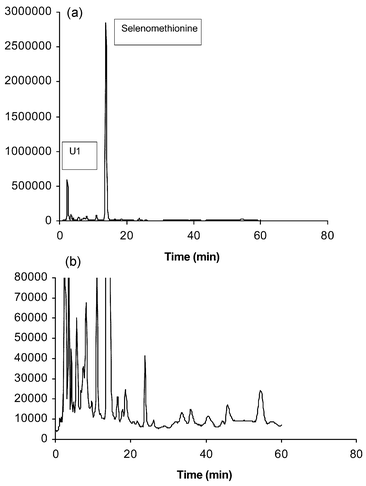 | ||
| Fig. 1 Se-specific HPLC-ICP-MS chromatograms of the enzymatic hydrolysis of 1250 ppm Se yeast (reference) using 0.1% HFBA ion pairing agent, Symmetry Shield™ column (lower—Se-82 counts s−1 response scale expanded). | ||
Replicate determinations made for multiple samples of SelenoExcell™ (n = 3 for each of 3 batches) produced chromatograms (as in Fig. 1) which could be overlaid and showed RSD values for the quantitation of identified components of 1% or better. Provided that the Protease XIV enzyme employed for digestion was kept at −40 °C when not in use, virtually no changes in chromatographic profiles were seen in six months. This ‘reference’ yeast reproducably showed ca. 13–14% of total chromatographable selenium eluted before selenomethionine and less than 1% eluted after that peak. Each time a new group of experiments were run or a new sample of Protease XIV was used the ‘reference’ yeast was determined to confirm consistent analytical behavior.
Table 2 shows the percentage compositions of the four previously identified selenium species obtained for a number of selenized yeasts with different total selenium contents. Total eluted selenium referenced against element content range from 69.2–94%, repeatability values being as noted above. Of this elutable selenium, the selenomethionine percentage ranged from 62.5% to 89.1%, selenite was always very low, but the Se-adenosylselenohomocysteine content varied more widely (0.5–7.4%). Unknown peaks U1 and, to a lesser extent, U2 were also present in the low percent range.
| Se-species (Table 1) | 1250 ppm yeast | 1204 ppm yeast | 1258 ppm yeast | 1406 ppm yeast | 1500 ppm yeast | 550 ppm yeast | 1200 ppm yeast | 1220 ppm yeast | 1150 ppm yeast |
|---|---|---|---|---|---|---|---|---|---|
| a %Se distributions are expressed in terms of total selenium content eluting from the column. Compounds with distribution percentage values marked with an asterisk were confirmed by HPLC-ESI-MS. | |||||||||
| 1 | 0.09% | 0.2% | 0.4% | 0.4% | 0.04% | 0.03% | 0.1% | 0.09$ | 0.5% |
| 12 | 84%a | 77.2% | 62.5% | 77.4% | 83.3% | 89.1% | 66.5% | 74.5% | 74%a |
| 15 | 0.54% | 0.9% | 1.6% | 1.2% | 0.9% | 0.1% | 0.9% | 0.9% | — |
| 18 | 0.5%a | 0.9% | 4.7% | 7.4% | 5% | 4.8% | 5.9% | 4.4% | — |
| SUM | 85.1% | 79.2% | 69.2% | 86.3% | 89.2% | 94% | 73.4% | 79.9% | 74.5% |
Selenium-enriched nutritional tablets19
Previously reported selenium speciation and quantitation showed markedly different results for selenium-enriched tablets obtained from various sources. Sutton et al. in particular23 noted a wide variation among supplement tablets. A typical supplement tablet weighs ca. 500 mg and contains 200 µg of selenium present in 160 mg of selenized yeast. The remaining excipients comprise a range of ingredients such as calcium phosphate, cellulose gel, stearic acid, silica, hydroxypropylcellulose, magnesium stearate and polyethylene glycol, but no specific antoxidants.As part of an on-going study related to the National Cancer Institute human intervention trials previously reported19 and proposed for the future,22 samples of selenium-enriched tablets used in the former trial were obtained from archived samples. Representative tablets were crushed, after first manually removing titanium dioxide coatings if present, and treated enzymatically according to the standard protocol. Tablets were chosen from various time periods of the trial from 1986–1993 and are designated N2, N5, N8 and N13. A more comprehensive survey of the results from this study will be published elsewhere. A representative HPLC-ICP-MS chromatogram is shown in Fig. 2 for sample N5, showing a striking difference from the selenized yeasts analyzed previously. While selenomethionine is still present, it occurs in a much lower proportion and ‘unknowns’ U1 and U2 now appear as the predominant components. In all archived tablets examined these three peaks predominate in various proportions. Table 3 shows representative quantifiation of four archived tablets. Each result is for replicate analyses for representative samples from 10 pooled tablets of each batch. Repeatability was as noted above for yeast samples.
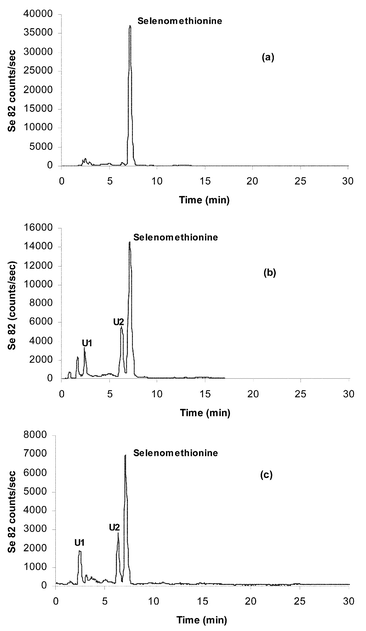 | ||
| Fig. 2 HPLC-ICP-MS chromatograms of the enzymatic hydrolysis of (a) SelenoExcell™ yeast, (b) Clark yeast and (c) Clark yeast tablet (N5), using 0.1% HFBA as ion-pairing agent. XTerra™ column | ||
| 1 | 2 | 12 | 15 | 18 | U-1 | U-2 | Sum | |
|---|---|---|---|---|---|---|---|---|
| a 1 = Selenate, 2 = selenite, 12 = selenomethionine, 15 = γ-glutamyl-Se-methylselenocysteine, 18 = Se-adenosylselenohomocysteine, U-1 = unknown 1, U-2 = unknown 2. Designations N2, N5, N8 and N13 are representative of sequentially numbered supplements administered in the ‘Clark study’, 1983–1993.22 | ||||||||
| N2 | — | 9.5 | 8.4 | — | — | 58.3 | 17.3 | 93.5 |
| N5 | — | 2.9 | 24.6 | 1.3 | 3.1 | 52.8 | 10.9 | 95.6 |
| N8 | — | 3.3 | 19.5 | — | 2.4 | 38.9 | 21.6 | 85.7 |
| N13 | — | 11 | 7.4 | — | 2.2 | 54.3 | 15.5 | 90.4 |
Since all of the archived tablets showed substantially different selenium profiles from the reference selenized yeast, it appears that changes in speciation (as determined following enzymatic treatment and extraction) have occurred, either from lengthy storage, perhaps by oxidation, by interaction with tablet excipients or perhaps by linked processes. An important observation suggests that excipients in tablets are not involved since a selenized yeast powder obtained from the University of Arizona archives designated as ‘Clark yeast’, which was used in the preparation of ‘N’ series samples, gave a profile very similar to that of N5 (Fig. 2). Fig. 2 also compares these profiles with that of reference yeast powder employed to prepare currently manufactured selenized yeast tablets (SelenoExcell™).
Oxidation of selenoamino acids
The selenoxides of selenoamino acids have been found in natural samples26–29 and, in the case of selenized yeasts or nutritional supplement extracts, selenium compounds eluting early under reversed phase mobile phase conditions, may be such oxidation products.Amino acid selenoxides have been synthetically produced and studied, primarily because of interest in the antioxidant activity of selenium. Gammelgaard et al. recently carried out an exhaustive study of the oxidative degradation of selenomethionine.30 These workers separated oxidation products during the course of the process and used ICP-MS, LC-MS and NMR to identify major compounds. Selenomethionine selenoxide and methaneseleninic acid were fully characterized.
We have previously reported the examination of oxidation of selenoamino acids by HPLC-ICP-MS and found differing products for different acids.11,12 In particular, small pH changes in HPLC separation conditions may give rise to data needing careful interpretation, especially as regards retention time shifts. Thus, while the primary product of peroxide oxidation of selenomethionine is selenomethionine selenoxide hydrate, oxidation of Se-methylselenocysteine generates methaneseleninic acid among other products.11,26 The treatment of SelenoExcell™ yeast with excess hydrogen peroxide was now investigated, and the major product identified as selenomethionine selenoxide hydrate elutes at ca. 2.5 min on the Symmetry Shield™ column. In addition, a small amount of selenite is present after the oxidation.
In the enzymatic extraction of SelenoExcell™ yeast two major components were found: selenomethionine and “U1” at ca. 2.5 min on the Symmetry Shield™ column (Fig. 1). Since “U1” elutes in the early part of the chromatogram it is considered to be hydrated selenomethionine selenoxide. This is minimally retained due to the enhanced polarity imparted by the addition of oxygen and the elements of water to the selenium atom, producing greater affinity for the acidic aqueous mobile phase. The formation of the hydrate under such acid conditions has been reported elsewhere.30,31 The mass spectrum of oxidized selenomethionine was confirmed as reported by Bird to indicate the presence of hydrated selenomethionine selenoxide with a selenium ion cluster (Se-80 molecular ion at m/z 232).32 Larsen et al. have described an ion exchange HPLC-ICP-MS study in which selenomethionine selenoxide was observed and confirmed.14 ‘U2’ is chromatographically retained similarly to selenomethionine and thus is considered to have a considerably less polar structure than the hydrate. Its retention is in the region typically seen for ion paired selenamino acids.
Reduction of oxidized SelenoExcell™
As was noted earlier,16 the oxidation of selenomethionine to selenomethionine selenoxide is reversible upon reduction by thiosulfate in aqueous solution, as shown by proton NMR.33 For selenomethionine standards, we confirmed earlier by HPLC-ICP-MS that quantitative recovery of non-oxidised selenomethionine occurs upon treating the hydrated selenomethionine selenoxide with excess thiosulfate.16Upon addition of thiosulfate to oxidized SelenoExcell™ yeast extract, the peak considered to be selenomethionine selenoxide hydrate reverted to selenomethionine, the chromatographic profile of which was, however, complicated by broadening and double peaking, the extent of which was dependent on the amount of thiosulfate added with resulting pH change. Such behavior was attributed to equilibrium between non-protonated and protonated/ion paired forms of selenomethionine.13 Coincidentally this chromatographic peak behavior overlapped with U2 and led initially to an erroneous conjecture that U2 corresponded to another ‘derivative’ of selenomethionine, perhaps the nonhydrated selenomethionine selenoxide. This supposition was, however, considered unlikely since at the pH of the HFBA mobile phase of 2.55, only the hydrated form would be expected.13 This behavior prevented the archived samples from being usefully subjected to the oxidation–reduction sequence as U2 was masked by the behavior of selenomethionine. The need for an alternative independent analytical procedure to elucidate U2 was clear.
Gas chromatography
Capillary gas chromatography provides sensitive, high-resolution analysis for analytes of sufficient volatility, or following chemical derivatization to convert non-volatile species to volatile ones. When coupled with atomic emission detection (AED), selenium in target molecules is directly monitored by spectroscopic detection of atomic emission radiation at a wavelength characteristic of the element, providing both qualitative elemental speciation and quantitation.34,35 We have used GC-AED to detect and determine many volatile organoselenium compounds present in or produced by plants such as garlic, elephant garlic, onion and broccoli.21 A common structure is R–Sx(Sey)–R′, where R and R′ are methyl or allyl groups.Derivatization with alkyl chloroformates provides an effective route to volatile species for amino acid GC. Selenoamino acids have been derivatized for GC with isopropyl chloroformate,36 and ethyl chloroformate; selenocysteine, Se-methylselenocysteine and selenomethionine have been determined in normal and selenium-enriched plants using GC-AED.37,38
Depicted in Fig. 3 is a GC-AED chromatogram (Se at 196 nm) of the ethyl chloroformate derivatives of six selenoamino acids, all of which elute within a seven minute window with excellent resolution and peak efficiencies. The retention times of these species are shown in Table 4. The longer retention time of the selenocysteine derivative results from the fact that derivatization takes place at the Se–H group as well as the amino and carboxylate functions.
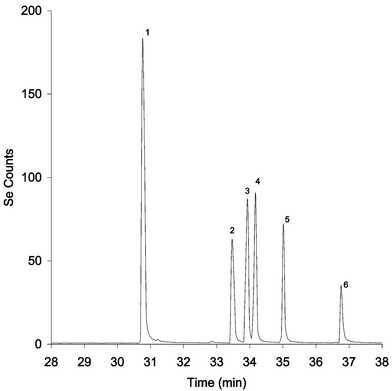 | ||
| Fig. 3 GC-AED temperature programmed chromatogram of derivatized selenoaminoacids. (1) Se-methylselenocysteine, (2) selenomethionine, (3) Se-allylselenocysteine, (4) Se-propylselenocysteine, (5) selenoethionine and (6) selenocysteine. | ||
| Name | Molecular weight | Retention time/min |
|---|---|---|
| Se-Methylselenocysteine derivative | 283 | 30.75 |
| Selenomethionine derivative | 297 | 33.52 |
| Se-Allylselenocysteine derivative | 309 | 33.95 |
| Se-Propylselenocysteine derivative | 311 | 34.20 |
| Selenoethionine derivative | 311 | 35.00 |
| Selenocysteine derivative | 341 | 36.75 |
Fig. 4 shows the GC-AED (Se at 196 nm) of derivatized Clark yeast and SelenoExcell™ yeast extract prepared by the Protease XIV digestion of yeast powders, and of derivatized selenomethionine as a reference. These were the same samples as investigated by HPLC-ICP-MS as described earlier. It is apparent (Fig. 4) that the Clark yeast chromatogram contains more peaks than the SelenoExcell™ or selenomethionine chromatograms. Particularly interesting is the peak appearing at ca. 35 min in the region corresponding to derivatized selenoamino acids. In Fig. 5 a comparison of selenium chromatograms for archived N2, N5 and N8 archived tablets prepared by Protease XIV digestion followed by derivatization are seen. The selenium-containing component at 35 min appears for each of the tablets to varying degrees.
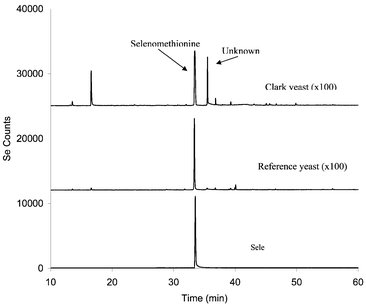 | ||
| Fig. 4 GC-AED (Se 196 nm) of derivatized selenomethionine, Clark yeast and SelenoExcell™ yeast. Baseline offset for display purposes: Reference yeast trace, baseline is at +12000 counts; Clark yeast trace, baseline is at +25000 counts. | ||
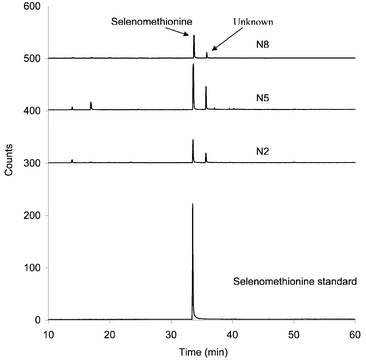 | ||
| Fig. 5 GC-(Se 196 nm) of N2, N5 and N8 Clark yeast tablets using the enzymatic extraction method and derivatization by ethyl chloroformate. Baseline offset for display purposes: N2 trace, baseline is at +300 counts; N5 trace, baseline is at +400 counts, N8 trace, baseline is at +500 counts. | ||
A key advantage of GC-AED is the facility to display simultaneously a number of element specific chromatograms for different elements. In this fashion, carbon, sulfur and selenium chromatograms can be displayed simultaneously. Fig. 6 shows comparative C, S and Se chromatograms for the Clark yeast. The selenium and sulfur traces indicate the presence of both of these elements in the peak eluting at ca. 35 min. Independent calibrations performed to estimate relative responses of S and Se under the set plasma conditions, using derivatized methionine and selenomethionine as standards,39 showed the selenium response at 196 nm to be approximately four times the sulfur response at 181 nm. Accordingly it was concluded from Fig. 6 that the species contained equal numbers of Se and S atoms. Further, given the retention time of the peak, only one of each atom could be present in the parent molecule, presumed to be a selenoamino acid. Additional strong evidence for the existence of both S and Se in the molecule was gained from spectral examination of the plasma emission present at this peak, the sulfur emission lines at 181, 182 and 183 nm being seen as well as the selenium 196 nm emission line.
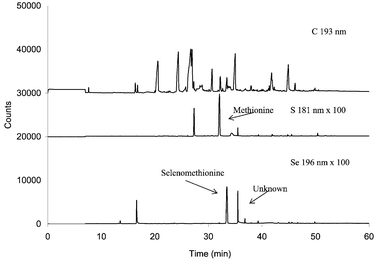 | ||
| Fig. 6 GC-AED chromatogram of derivatized Clark yeast using the enzymatic extraction and treatment with ethyl chloroformate. | ||
Other evidence was gathered to shed light on the possible nature of this 35 min peak, which was conjectured at this stage to be produced by derivatization of the peak denoted U2 in the liquid chromatograms. Thus, treatment of the Clark yeast extract with 50 mM dithiothreitol, a reagent established to cleave S–S or Se–Se bonds, removed the 35 min peak from the GC-AED chromatogram while leaving the derivatized selenomethionine peak untouched. A similar experiment performed using HPLC-ICP-MS analysis gave the same result. Additionally the U2 peak was carefully trapped from the HPLC column for a number of injections and concentrated by freeze-drying. After derivatization this fraction also showed the 35 min peak in S and Se channels by GC-AED (Fig. 7).
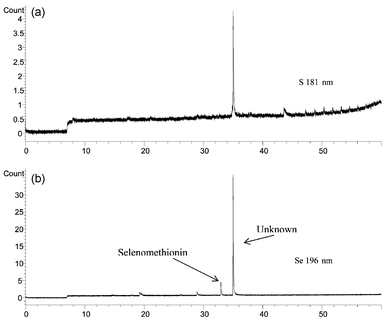 | ||
| Fig. 7 GC-AED (Se 196 nm and S 181 nm) of trapped fraction collected from HPLC of Unknown 2, derivatized by ethyl chloroformate. | ||
Evidence was now growing that U2 was an amino acid containing a Se–S bond. Such was the low level of the species present and the preponderance of other co-eluting non-selenium species in the HPLC present in the proteolytically extracted yeast that direct HPLC-mass spectrometry proved to be impossible with the available instrumentation. Accordingly, the ethylated derivative was examined by gas chromatography-mass spectrometry.
Fig. 8 shows the total ion chromatogram of the derivatized extract and the mass spectrum obtained at the peak eluted at a retention time corresponding to the 35 min peak in GC-AED. The main features of the spectrum were readily interpreted as follows. The molecular ion peak (M+) is seen at m/z 315 (80Se) and it also showed the selenium isotope pattern. The cluster of ions at m/z 242 suggests the selenium isotope pattern and loss of COOCH2CH3 (m/z 73) from the molecular ion (M+). The prominent ion at m/z 220 indicates the loss of SeCH3 (m/z 95) and shows no selenium isotope pattern. The ion at m/z 188 may be interpreted as the loss of SSeCH3 (m/z 127) and that at m/z 174 as due to the loss of CH2SSeCH3 (m/z 141). This spectrum gives a strong indication that the U2 molecule contained the CH2SSeCH3 moiety and was also an amino acid. The conjectured acid is thus S-(selenomethyl)cysteine, CH3SeSCH2CH(NH2)COOH, a previously unreported species.
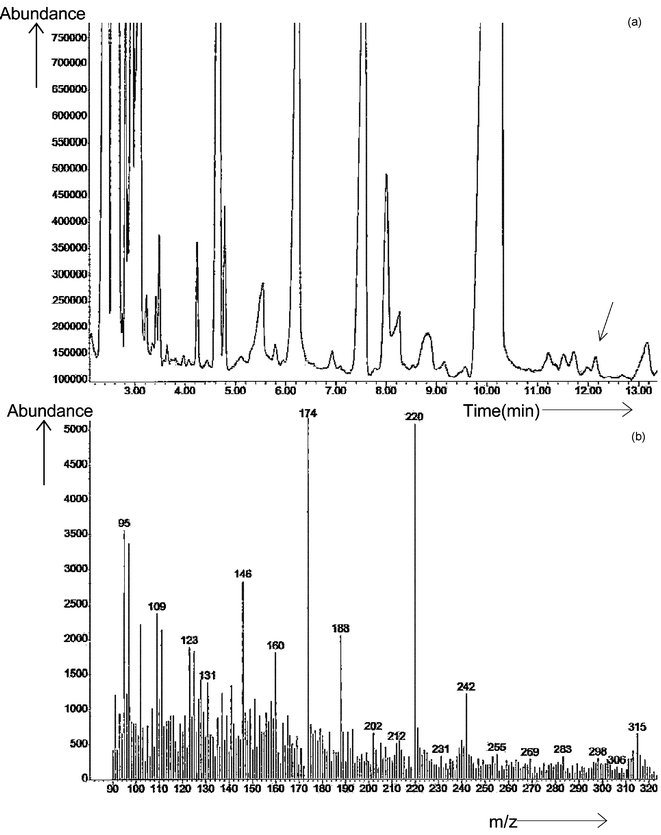 | ||
| Fig. 8 (a) GC-MS total ion chromatogram of Clark yeast using the extraction method and derivatized by ethyl chloroformate; (b) mass spectrum of the unknown peak U2 in Clark yeast, molecular ion m/z = 315 (80Se). | ||
While it was considered that there was good evidence for this assignment, the independent synthesis of this molecule was accomplished and will be reported elsewhere.40 This synthetic sample was derivatized by the standard method and showed the GC-MS characteristics in Fig. 9. In this spectrum, the principal features are much simpler to observe since the ion background is much lower. The principal ions at m/z 315, 242, 220, 188 and 174 appear clearly. There are possible assignments based upon apparent selenium isotope patterns of fragments CH2SSeCH3 (m/z 141) and SeCH3 (m/z 95). There is a direct correspondence with the U2 derivative and it is considered that the evidence of identification is conclusive.
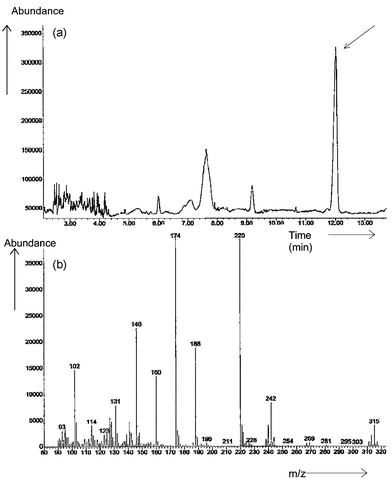 | ||
| Fig. 9 (a) GC-MS total ion chromatogram of synthesized U2, derivatized by ethyl chloroformate; (b) mass spectrum of selenomethylcysteine, molecular ion m/z = 315 (80Se). | ||
Conclusion
High-selenium yeast in both free yeast and tableted forms, stored at room temperature for more than ten years, gives rise, after subjection to proteolytic digestion and extraction, to substantial amounts of selenomethionine selenoxide hydrate and the previously unreported selenoamino acid S-(selenomethyl)cysteine. These observations have important nutritional and perhaps metabolic implications. The species now identified as selenomethionine selenoxide hydrate is also observed in smaller proportions for some contemporary selenized yeasts and it is reasonable to conclude that oxidation of selenomethionine in the presence of water contributes variously to this finding in different samples. From the standpoint of nutritional efficacy and selenium uptake, it is possible that the selenoxide is equivalent in action to selenomethionine and is metabolized in the same way. The inclusion of the selenoxide in quantitative measurement of selenomethionine content for selenized yeast seems reasonable. The existence of S-(selenomethyl)cysteine has broader import that will be examined further. Perhaps the presence of this Se–S species may be related to as yet undefined selenium metabolism. It is not known if speciation changes can occur over the shorter time that the product might be held in storage, but closer examination of other current yeast products suggests that a previously unidentified component present at low concentrations may be S-(selenomethyl)cysteine. Studies using the Se-fingerprint method to evaluate the effects of accelerated aging (i.e., high-temperature exposure) on the pattern of Se-compounds in selenized yeast are clearly important. Combs et al.41 recently reported the direct measurement of the hitherto unrecognized fraction of plasma containing low molecular weight (<5 kD) Se compounds. This is an important finding; inasmuch as it indicates the presence in plasma of one or more Se-species that, if they can be characterized, may offer new and more informative ways to assess Se metabolic status.Acknowledgements
This material is based upon work supported by the National Science Foundation under Grants No. 0094568 (J.F.T) and CHE-9906566 (E.B.) and the Petroleum Research Fund, administered by the American Chemical Society (E.B.). Samples and support from Philip Taylor, MD, NIH-NCI, Cypress Systems and McKesson Biosystems are acknowledged. We thank the PerkinElmer Corporation for provision of the Elan 5000 plasma source mass spectrometer. The provision of XTerra™ columns by Waters Chromatography Corporation is gratefully acknowledged. The authors also thank Thomas Houle and Mount Holyoke College for providing access to GC-MS instrumentation.References
- O. Wada, N. Kurihara and N. Yamazaki, Jap. J. Nutr. Assess., 1997, 10, 199 Search PubMed.
- H. E. Ganther, Carcinogenesis, 1999, 20(9), 1657 CrossRef CAS.
- O. A. Levander, Ann. Rev. Nutr., 1987, 7, 227 CrossRef CAS.
- C. Ip, M. Birringer, E. Block, J. F. Kotrebai, J. F. Tyson, P. C. Uden and D. J. Lisk, J. Agric. Food Chem., 2000, 48, 2062 CrossRef CAS.
- G. F. Combs and W. P. Gray, Pharmacol. Ther., 1998, 79, 179 CrossRef CAS.
- H. E. Ganther and J. R. Lawrence, Tetrahedron, 1997, 53, 12229 CrossRef CAS.
- A. D'Ulivo, Analyst, 1997, 122, 117R RSC.
- K. Pyrzynska, Analyst, 1996, 121, 77R RSC.
- R. Lobinski, J. S. Edmonds, K. T. Suzuki and P. C. Uden, Pure Appl. Chem., 2000, 72, 447 Search PubMed.
- P. C. Uden, Anal. Bioanal. Chem., 2002, 373, 422 CrossRef CAS.
- M. Kotrebai, J. F. Tyson, E. Block and P. C. Uden, J. Chromatog., 2000, 866, 51 CrossRef CAS.
- P. C. Uden, S. M. Bird, M. Kotrebai, P. Nolibos, J. F. Tyson, E. Block and E. R. Denoyer, Fresenius J. Anal. Chem., 1998, 362, 447 CrossRef CAS.
- N. Jakubowski, D. Stuewer, D. Klockow, C. Thomas and H. J. Emons, J. Anal. At. Spectrom., 2001, 4E Search PubMed.
- H. Larsen, M. Hansen, T. Fan and M. Vahl, J. Anal. At. Spectrom., 2001, 16, 1403 RSC.
- S. McSheehey, P. Pohl, J. Szpunar, M. Potin-Gautier and R. Lobinski, J. Anal. At. Spectrom., 2001, 16, 68 RSC.
- M. Montes-Bayon, D. L. LeDuc, N. Terry and J. A. Caruso, J. Anal. At. Spectrom., 2002, 17, 872 RSC.
- X.-J. Cai, P. C. Uden, E. Block, X. Zhang, J. J. Sullivan and B. D. Quimby, J. Agric. Food Chem., 1994, 42, 2085 CrossRef.
- X.-J. Cai, E. Block, P. C. Uden, J. J. Sullivan and B. D. Quimby, J. Agric. Food Chem., 1995, 43, 1751 CrossRef CAS.
- L. C. Clark, G. F. Combs, B. W. Turnbull, E. H. Slate, D. K. Chalker, J. Chow, L. S. Davis, R. A. Glover, G. F. Graham, E. G. Gross, A. Krongrad, J. L. Lesher, K. Park, B. B. Sanders, C. L. Smith and R. Taylor, J. Am. Med. Assoc., 1996, 276, 1957 Search PubMed.
- C. Ip, Z. Chu, H. J. Thompson, D. J. Lisk and H. E. Ganther, Anticancer Res., 1999, 19, 2875 Search PubMed.
- C. Ip, J. Nutr., 1998, 128, 1845 CAS.
- E. A. Klein, I. M. Thompson, S. M. Lippman, P. J. Goodman, D. Albanes, P. R. Taylor and C. Coltman, World J. Urol., 2003, 21, 21 Search PubMed.
- K. L. Sutton, C. A. Ponce de Leon, K. L. Ackley, R. M. C. Sutton, A. M. Stalcup and J. A. Caruso, Analyst, 2000, 125, 281 RSC.
- S. McSheehy, F. Pannier, J. Szpunar, M. Potin-Gautier and R. Lobinski, Analyst, 2002, 127, 223 RSC.
- P. Husek, J. Chromatogr., 1991, 552, 289 CrossRef CAS.
- E. Block, M. Birringer, W. Jiang, T. Nakahodo, H. J. Thompson, P. J. Toscano, H. Uzar, X. Zhang and Z. Zhu, J. Agric. Food Chem., 2001, 49, 458 CrossRef CAS.
- M. Rooseboom, J. N. Commandeur, G. C. Floor, A. E. Rettie and N. P. Vermeulen, Chem. Res. Toxicol., 2000, 14, 127 CrossRef CAS.
- J. E. Cone, R. M. Del Rio, J. N. Davis and T. C. Stadtman, Proc. Natl. Acad. Sci. USA, 1976, 73, 2659 CAS.
- S. Padjama, G. L. Squadrito, J. N. LeMercier, R. Cueto and W. A. Pryor, Free Rad. Biol. Med., 1996, 21, 317 CrossRef CAS.
- B. Gammelgaard, C. Cornett, J. Olsen, L. Bendhal and S. H. Hansen, Talanta, 2003, 59, 1165 CrossRef CAS.
- H. A. Zainal, D. E. Lacroix and W. R. Wolf, Fresenius’ J. Anal. Chem., 1996, 355, 311.
- S. M. Bird, PhD Dissertation, University of Massachusetts, 1998.
- A. A. Isab, Inorg. Chim. Acta, 1983, 80, L3 CrossRef CAS.
- B. D. Quimby, P. C. Uden and R. M. Barnes, Anal. Chem., 1978, 50, 2112 CrossRef CAS.
- P. C. Uden, Element-Specific Chromatographic Detection by Atomic Emission Spectroscopy,, American Chemical Society Symposium Series #479, ACS, Washington, DC, 1992 Search PubMed.
- H. Kataoka, Y. Miyanaga and M. Makita, J. Chromatogr. A, 1994, 659, 481 CrossRef CAS.
- X.-J. Cai, E. Block, P. C. Uden, X. Zhang, J. J. Sullivan and B. D. Quimby, J. Agric. Food Chem., 1995, 43, 1754 CrossRef CAS.
- K. Yasumoto, T. Suzuki and M. Yoshida, J. Agric. Food Chem., 1988, 36, 463 CrossRef CAS.
- P. Nolibos, PhD Dissertation, University of Massachusetts, 2001.
- E. Block, R. S. Glass, S. Johnson, H. Totoe Boakye, C. Kahakachchi, J. F. Tyson and P. C. Uden, J. Agric. Food Chem. Search PubMed , submitted for publication.
- G. F. Combs, Jr., T. Hyum and W. P. Gray, in Metal Ions in Biology and Medicine, eds. J. A. Centeno, P. Collery, G. Verney, R. B. Finkelmen, H. Gibb and J. C. Etienne, John Libbey Eurotext, Paris, 2000, pp. 237–240 Search PubMed.
| This journal is © The Royal Society of Chemistry 2004 |