A green look at the aldol reaction
Ramon
Mestres
Departament de Química Orgànica, Universitat de Valencia, Dr. Moliner 50, 46100, Burjassot, València, Spain. E-mail: ramon.mestres@uv.es; Fax: + 34 93 588 84 03; Tel: + 34 93 588 84 03
First published on 5th November 2004
Abstract
Aldol reactions have been and are widely applied for the preparation of β-hydroxy aldehydes, β-hydroxy ketones or α,β-unsaturated aldehydes or ketones through addition or addition-elimination reactions of aldehydes and ketones. The study of the aldol reaction from the point of view of its greenness must have in mind first of all that a general synthetic method must be based on complete and efficient conversions of well defined selectivity and that greenness is more a term for comparison than an absolute kind of qualification. This comparison, when referred to the aldol reaction, applies here to the diverse modifications of the reaction. Thus, the original poorly selective, but highly atom economic catalytic procedures, have been improved by several authors by introduction of stoichiometric forms of activation in the search for better selectivities. However, the success in these improvements has been accompanied by higher levels of hazard and waste. The study of the greenness of the aldol reaction is completed by a short overview of recent contributions intended to achieve efficient, safe and clean conversions that are susceptible to becoming general green synthetic procedures. Interesting contributions are highlighted for reactions carried out under solvent-less conditions, in water, ionic liquids and supercritical fluids, with activation by microwaves, or with use of heterogeneous catalysis and especially of biocatalysis and biomimetic catalysis. Promising methods based on reduction of unsaturated ketones or on rearrangement of allylic alcohols have also recently been described.
 Ramon Mestres Ramon Mestres | Ramon Mestres was born in Barcelona, Spain, in 1937. He received his PhD in Chemistry from Universidad de Barcelona in 1962 (thesis under Prof Dr José Pascual) and his DPhil in Chemistry from University of Oxford in 1965 (thesis under Sir Ewart RH Jones). In 1966 he joined Universidad de Navarra. Currently he is Full Professor of Organic Chemistry in Universitat de Valencia, Spain and Honorary Professor of Universidad de Piura, Peru. He is Chairman of the Green Chemistry Committee in the FECS Division for Chemistry and the Environment and Chairman of the Spanish Green Chemistry Network. His current research interests are: reactivity of dienediolates of carboxylic acids, reaction mechanisms and green organic synthetic methods. |
Introduction
The aldol reaction is one of the most powerful and best known C–C forming synthetic reactions, universally present in basic and advanced organic chemistry texts and amply reviewed in organic synthetic books and series.1–4 The reaction has industrial relevance either in bulk production, or in the fine chemical and pharmaceutical industry.5 The aldol reaction has a particular interest in that it may be qualified as one of the natural synthetic methods in the sense that important biogenetic pathways are based on aldol conversions and its application opens access to highly functionalised natural and non-natural substances, related for instance, to carbohydrates. The aim of this article is to present a first green view of the well established and current methods of the aldol reaction and to introduce recent contributions, some of them susceptible to becoming general green synthetic procedures.In order to evaluate the greenness of a particular process attention must be paid in the first instance to issues related to safety, health and protection of the environment, due to reactants (substrates and reagents), auxiliaries (mainly solvents) and waste. This enumeration is obviously incomplete, but can be useful at present. The question about how green a reaction is most frequently refers to a particular conversion, to the comparison between two or more alternative processes for the same synthetic target, or between the synthetic pathways for the manufacture of alternative compounds. Several approaches based on the twelve principles of Green Chemistry formulated by Anastas and Warner and on the twelve more green principles due to Winterton have been developed in order to give an adequate answer to that question.6–8 However, the present study deals with general procedures and a slightly different approach is needed. Indeed, comparison can now be done between reagents, catalysts and solvents, not with particular substances, but rather with classes of compounds. This comparison should show to what extent synthetic features achieved by diverse procedures are paralleled by modifications in hazard and production of waste.
General properties of substrates, reagents, solvents and waste, especially their toxicity and their connection to the sources of industrial bulk chemicals will be taken into account. Atom economy of the present general procedures will be based on the stoichiometry of the reactions and defined through molar waste of the reaction. Thus, grams of concomitants for each mole of product will be used here, instead of the generally accepted atomic yield or the E-Factor, which is more convenient if particular substances are involved.9 Harmless and ecologically non-significant concomitants, namely water, nitrogen, or oxygen or alkaline, and alkaline earth metal salts in catalytic amounts are not taken into account in the estimation of molar waste. Solvents are assumed to be recovered, although risks associated with their use or their susceptibility to contribute to contamination must be considered.
The aldol reaction
Aldol reaction is here understood in its strict sense of a C–C forming reaction which involves aldehydes and ketones as reactants. However currently developed catalytic aldol producing reactions which start from α,β-unsaturated ketones or from allylic alcohols as precursors of the enolates will be briefly introduced because of their green significance. The related Knoevenagel, Reformatsky, Perkin, Mannich, Stobbe, or Darzens reactions and other conversions of carbonyl compounds with nitroalkanes, esters, or amides will be studied elsewhere. Additionally, only intermolecular aldol reactions will be considered here.Selectivity
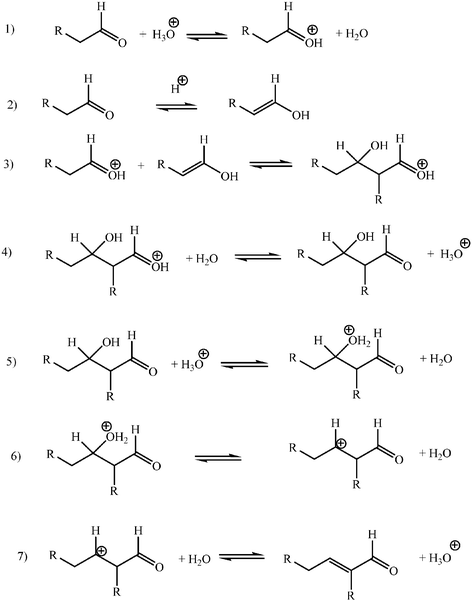
A general synthetic method, especially when intended to be environmentally benign, should be based on complete conversions of well defined selectivity. Inversion of selectivity trends at will would be desirable. It may be convenient then to have a look at the structural possibilities for selectivity that the aldol reaction offers. Aldol reactions lead to β-hydroxy aldehydes (aldols) or to β-hydroxy ketones (ketols) through an addition reaction (aldolization) or to the α,β-unsaturated aldehydes or ketones that result from a subsequent dehydration (aldol condensation) (Scheme 1, eqns 1 and 2). The reaction may involve two molecules of the same aldehyde or ketone (self-aldolization or self-condensation) or two different substances (cross-aldolization or cross-condensation). In any case, one of the molecules reacts as a carbonacid (donor) that adds to the carbonyl group of the other one (acceptor). In cross-reactions two donor–acceptor combinations are possible, unless one of the reactants has no α-hydrogen atoms (eqns 3 and 4). Condensations require two hydrogen atoms at the α-carbon atom of the donor aldehyde or ketone (eqn 5). Asymmetric ketones with α and α′ hydrogen atoms can react as donors to give two different ketols (eqns 6 and 7), or two enones when dehydration is possible.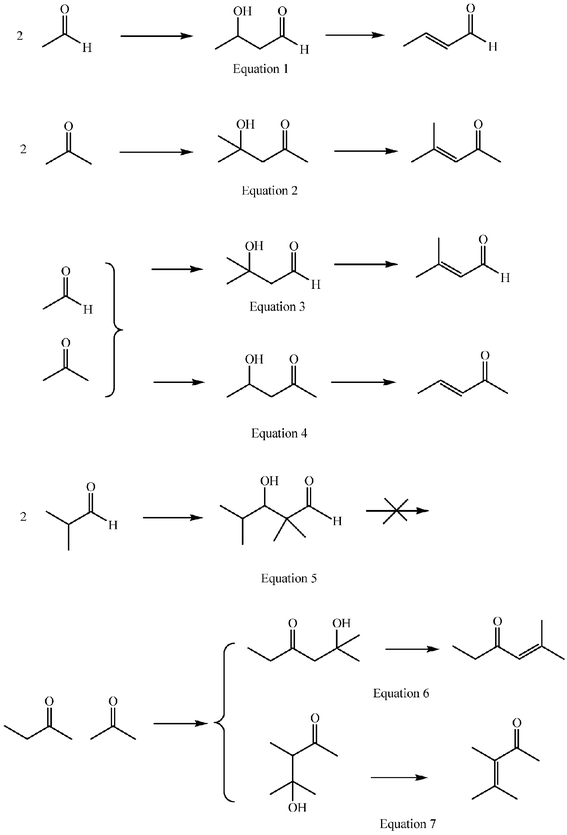 | ||
| Scheme 1 | ||
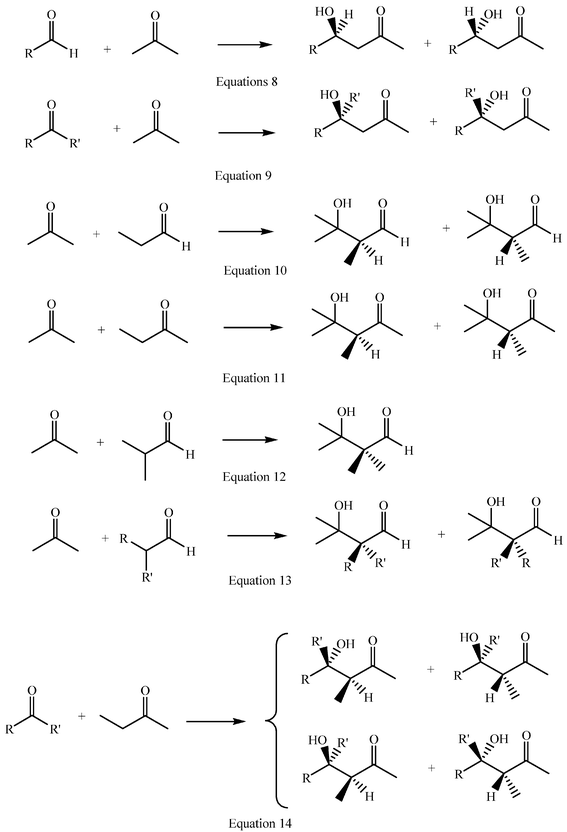
Aldehydes, other than formaldehyde, and asymmetric ketones have a prochiral carbonyl group which becomes chiral whenever they react as acceptors (Scheme 2, eqns 8 and 9). The same is true for α-methylene groups of aldehydes or ketones when they react as donors (eqns 10 and 11). α-Methyne groups are either chiral or achiral, but not prochiral, and their topicity is not modified on reaction as carbonacid centres (eqns 12 and 13). When a single stereogenic centre is generated or preserved aldol additions lead to a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) ∶1 mixture of a pair of enantiomers (eqns 8 to 13), unless the reaction becomes enantioselective. If the addition involves two prochiral centres, two enantiomeric pairs of erythro/threo
(or syn/anti) diastereoisomers may result and the addition is then susceptible to becoming both diastereoselective and enantioselective (eqn 14). The presence of other chiral centres in the reactants modifies the above situations, but its discussion is beyond the present purpose. On dehydration of aldols or ketols, C–C double bonds form and mixtures of cis/trans
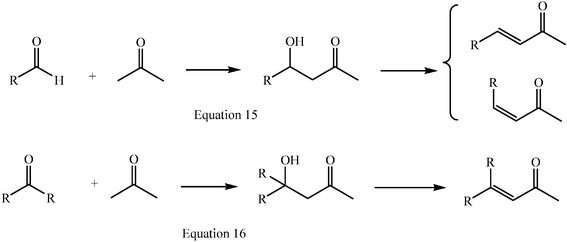
(E/Z) diastereoisomers result, except when the acceptor is either formaldehyde or a symmetrical ketone, or when the reaction is E- or Z-selective (Scheme 3, eqns 15 and 16).
∶1 mixture of a pair of enantiomers (eqns 8 to 13), unless the reaction becomes enantioselective. If the addition involves two prochiral centres, two enantiomeric pairs of erythro/threo
(or syn/anti) diastereoisomers may result and the addition is then susceptible to becoming both diastereoselective and enantioselective (eqn 14). The presence of other chiral centres in the reactants modifies the above situations, but its discussion is beyond the present purpose. On dehydration of aldols or ketols, C–C double bonds form and mixtures of cis/trans
(E/Z) diastereoisomers result, except when the acceptor is either formaldehyde or a symmetrical ketone, or when the reaction is E- or Z-selective (Scheme 3, eqns 15 and 16).
 | ||
| Scheme 2 | ||
 | ||
| Scheme 3 | ||
Aldol reactions were first carried out under simple general basic or acid homogeneous or heterogeneous catalytic conditions. Stoichiometric methods have been developed since the 1950s in order to overcome limitations in the scope and selectivity of those catalytic reactions. In these stoichiometric methods the donor is quantitatively deprotonated and the resulting enolate used directly or after conversion into a silyl enol ether or a boron enolate. A return to catalytic procedures has occurred recently in the search for asymmetric and for atom economically efficient procedures. This historical outline provides an adequate scheme for the present discussion.
Safety and environmental impact
Aldehydes and ketones are the starting materials in all procedures based on aldol reactions. It is convenient thus to introduce some comments on these classes of compounds before discussing procedures and reagents.A great number of simple starting aldehydes and ketones are bulk products and can be considered proximate to the fossil feed-stocks in the synthetic chain. Without going into detail, this feature allows the assumption that few reagent and energy consuming and waste producing conversions are needed for their manufacture. Other aldehydes and ketones are readily available natural products or can be easily prepared from them.
As for hazards associated with manipulation of aldehydes and ketones, it should be recalled here that all low molecular weight aldehydes are toxic. Formaldehyde and acetaldehyde are irritating to eyes, skin and tracheal tract and, more importantly, they have been reported as carcinogens. Saturated ketones are only moderately toxic, except on prolonged inhalation, when depression of the central nervous system may occur. One of the serious hazards associated with ketones is their ability to react with hydrogen peroxide to give dangerous explosive peroxides, a feature which, in connection with aldol reactions, should be cause for concern only in the work-up of boron enolate aldol reactions (see below).10 The above comments apply also to the aldol products: hydroxy aldehydes or ketones or their related unsaturated compounds. Thus, crotonaldehyde and methyl vinyl ketone are both included in EPA's “Extremely Hazardous Substances List” and acetaldol, and diaceton alcohol are reported as moderately irritant, toxic on skin contact and narcotic on inhalation.10
General catalytic aldol reactions
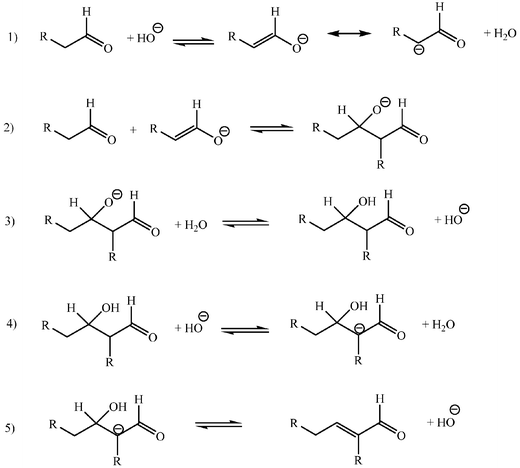
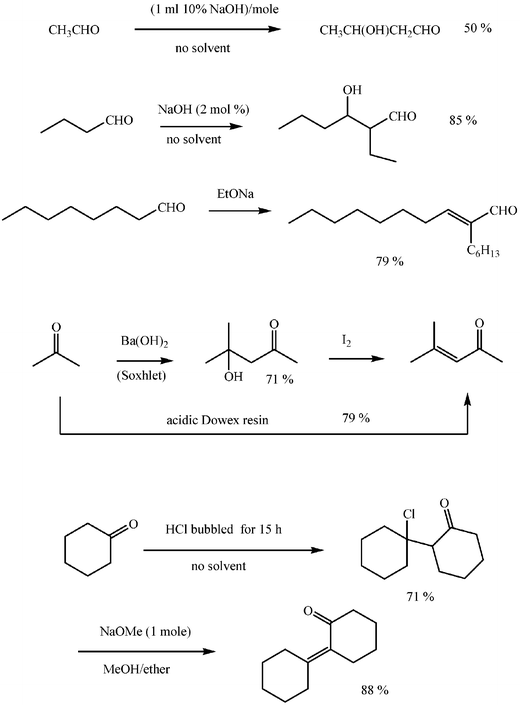
Typical bases for reactions carried out under basic catalytic conditions are alkaline or alkaline earth metal hydroxides. The use of basic ion-exchange resins as catalysts may be advantageous. Hydrochloric acid is the most frequent acid catalyst. Alcohols or alcohol–water systems are commonly used as solvents, although low molecular weight aldehydes and ketones react conveniently without solvent.2–5All the steps leading to aldolization are reversible (Schemes 4 and 5) and equilibrium constants may not be favourable to the progress of the conversion.11 Recognition of these features is crucial in order to understand reactivities and selectivities and to establish convenient experimental conditions. In general terms steric compression is detrimental for the addition, a feature that accounts for the poor reactivity of ketones as acceptors. Elimination steps are also reversible in theory, but frequently irreversible in practice, especially for acid catalysed reactions, and favoured when the forming ethylenic double bond is conjugated to a phenyl or another unsaturated group. The success of the conversion may depend occasionally on the continuous separation of products from the reacting mixture as they form, or on the practical irreversibility of the dehydration step. Good examples of these are the traditional laboratory scale aldol self-condensation of acetone in a Soxhlet set-up12 or the Claisen–Schmidt condensations carried out in water or in aqueous ethanol, frequently accompanied by precipitation of the unsaturated aldol adduct.
 | ||
| Scheme 4 | ||
 | ||
| Scheme 5 | ||
Linear alkyl aldehydes undergo self-aldolizations (Scheme 6), but only a limited number of ketones afford practical conversions. Control of pH and temperature of the solution is critical for the progress of the reaction to ensure that either addition or condensation results and to avoid oligomerizations, especially of condensation products. Bases are usually employed in catalytic amounts: 2% or 10% molar ratios are usual. This situation is not the same for acid catalysed reactions when about 2–3 equivalents of hydrochloric acid are frequently employed.3
 | ||
| Scheme 6 | ||
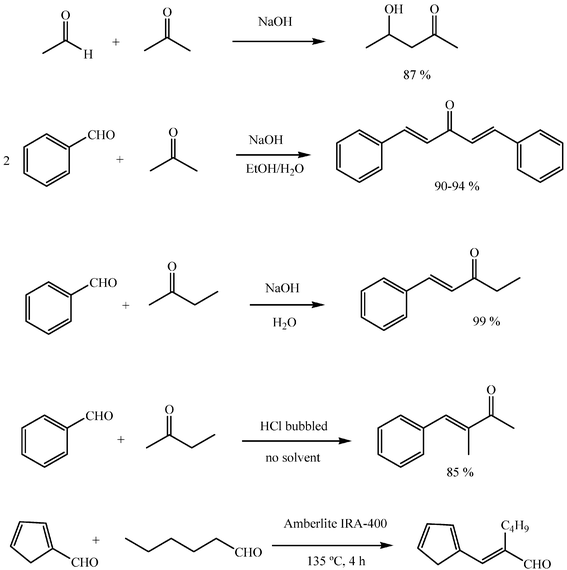
The cross reaction of two aldehydes affords a mixture of up to four compounds, which is simplified when one of them cannot participate in the reaction other than as an acceptor, or when different reactivities as acceptors allow one of the possible combinations to predominate in the mixture. Excess of the component which cannot allow self-condensation is then a common practice. A general application of cross aldol condensation is the condensation of aromatic aldehydes with other aldehydes or with ketones, known as the Claisen–Schmidt condensation (Scheme 7).
 | ||
| Scheme 7 | ||
Aldol reactions of asymmetric ketones may be regioselective. For instance, methyl ketones such as butanone and 2-pentanone react with a variety of aldehydes through their methylene groups. For the Claisen–Schmidt condensation, the general observation is that asymmetric ketones react with the aromatic aldehyde through the less substituted position (a methyl group in practice) under basic conditions and through the more substituted α position (a methylene group) under acid catalysis (Scheme 7).
The reversibility of most additions accounts for aldolizations not being stereoselective. However, trans geometry is frequently found in the condensation products of aldehydes with acetaldehyde or with methyl ketones, especially in the Claisen–Schmidt condensation.
Frequent side products found in aldol reactions are the carboxylic acids and alcohols (Cannizzaro) or esters (Tischenko) that result from disproportionation of the reacting aldehydes.2 The accompanying Cannizzaro reaction is frequently encountered in the use of formaldehyde and has industrial significance in the synthesis of pentaerythritol and related products.5 However, the oligomers that form by further reaction of the aldol products are probably the most important side products of catalytic aldol reactions.
Safety and environmental impact
In regard to safety and to environmental impact of catalytic aldol reactions, the easily available bases and acids are well known to chemists for their corroding properties, especially to human tissues, and their propensity to cause violent reactions on contact with a great variety of substances. Usual solvents, ethanol and methanol, are moderately flammable and methanol is accumulatively toxic.10 Their volatility may be a cause of concern and isopropyl alcohol stands as a likely substitute for both.7When attention is addressed to waste, not much should be expected, as long as promoter bases and acids are employed in genuinely catalytic amounts. Atom yield for carbon compounds in aldolization is 100% and only water is split in condensations. Work-up neutralization will lead then to small amounts of alkaline chlorides. Stoichiometric equations justify the assumption of a negligible molar waste for the basic catalysed aldol reaction (Table 1). Still better, ion exchange resins are easily separated and reused. For acid catalysis, the acid must be neutralized in the work-up and the resulting amount of alkaline chloride will depend on the actual amount of catalyst employed.
| Method | Stoichiometric equations | Concomitant products | Molar waste/g mol−1 |
|---|---|---|---|
| General base catalysis | 2CH3CHO + NaOH (cat. am.) → CH3CH(OH)CH2CHO + NaOH (cat. am.) | NaCl (cat. am.) | ≈0 |
CH3CH(OH)CH2CHO + NaOH (cat. am.)
→ CH3CH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CHCHO + H2O + NaOH (cat. am.) CHCHO + H2O + NaOH (cat. am.) |
|||
| NaOH (cat. am.) + HCl (cat. am.) → NaCl (cat. am.)+ H2O | |||
| Stoichiometric enolate generation | CH3COCH3 + i-Pr2NLi → CH3COCH2 Li + i-Pr2NH | i-Pr2NH | 101 |
| CH3COCH2 Li + CH3COCH3 → CH3COCH2–C(CH3)2OLi | LiCl | +42.5 | |
| CH3COCH2–C(CH3)2OLi + HCl → CH3COCH2–C(CH3)2OH + LiCl | =143.5 | ||
| For NaH as base | H2 | ||
| NaCl | 58.5 | ||
| Wittig aldol reaction | RCH2CHO + H2NC6H11
→ RCH2CHNC6H11
+ H2O |
i-Pr2NH | 101 |
| RCH2CHNC6H11
+ i-Pr2NLi →
[RCHCHNC6H11]Li +
i-Pr2NH |
LiCl | +42.5 | |
| [RCHCHNC6H11]Li + R′2CO → R′2C(OLi)CH(R)CHNC6H11 |
C6H11NH2 | +99 | |
| R′2C(OLi)CH(R)CHNC6H11
+ HCl → R′2C(OH)CH(R)CHNC6H11
+
LiCl |
=242.5 | ||
| R′2C(OH)CH(R)CHNC6H11
+ H2O + HCl (cat)
→ R′2C(OH)CH(R)CHO +
H2NC6H11 |
|||
| Reaction through silyl enol ether with regeneration of enolate | RCOCH3
+ i-Pr2NLi → RC(OLi)CH2
+
i-Pr2NH |
i-Pr2NH | 101 |
| RC(OLi)CH2
+
(CH3)3SiCl → RC[OSi(CH3)3]CH2
+
LiCl |
LiCl | +42.5 | |
| RC[OSi(CH3)3]CH2
+ CH3Li → RC(OLi)CH2
+
(CH3)4Si |
(CH3)4Si | +88 | |
| RC(OLi)CH2
+ R′2CO → RCOCH2C(OLi)R′2 |
+42.5 | ||
| RCOCH2C(OLi)R′2 + HCl → RCOCH2C(OH)R′2 + LiCl | =274 | ||
| For generation of silyl enol ethers under thermodynamic control | |||
| RCOCH3
+ Et3N +
(CH3)3SiCl → RC[OSi(CH3)3]CH2
+
Et3NH Cl |
Et3NH Cl | 123.5 | |
| (CH3)4Si | +88 | ||
| LiCl | +42.5 | ||
| =254 | |||
| Mukaiyama aldol reaction | RCOCH3
+ i-Pr2NLi → RC(OLi)CH2
+
i-Pr2NH |
i-Pr2NH | 101 |
| RC(OLi)CH2
+
(CH3)3SiCl → RC[OSi(CH3)3]CH2
+
LiCl |
LiCl | +42.5 | |
| RC[OSi(CH3)3]CH2
+ R′2CO + TiCl4
→ RCOCH2C(OTiCl3)R′2
+
(CH3)3SiCl |
(CH3)3SiCl | +108.5 | |
| RCOCH2C(OTiCl3)R′2 + H2O → RCOCH2C(OH)R′2 + Ti(OH)4 + 3HCl | Ti(OH)4 | +116 | |
| 3HCl + 3NaOH → 3NaCl + 3H2O | 3NaCl | +175.5 | |
| =543.5 | |||
| Boron enol ether aldol reaction | RCOCH3
+
(C4H9)2BOSO2CF3
+
(C2H5)3N → RC(OBBu2)CH2
+
(C2H5)3NH OSO2CF3 |
(C2H5)3NH OSO2CF3 | 251 |
| RC(OBBu2)CH2
+ R′2CO → RCOCH2C(OBBu2)R′2 |
+148 | ||
| RCOCH2C(OBBu2)R′2 + H2O2 → RCOCH2C(OH)R′2 + 2C4H9OH + B(OH)3 + H2O | 2C4H9OH | +62 | |
| B(OH)3 | =461 | ||
In view of the above comments, it may be concluded that the catalytic aldol reaction can be regarded as a substantially waste free method, especially when carried out in the presence of catalytic amounts of base or with basic ion-exchange resins. But there are significant limitations as a synthetic method. For instance, it does not provide adequate control of cross reactions; it does not allow the synthesis of the aldols or ketols which should result from ketones as acceptors; there is no convenient regioselectivity control of asymmetric ketones as donors; aldolizations are not stereoselective and the cis/trans stereochemistry of aldol condensation products cannot be controlled at will.
Stoichiometric aldol reactions
Stoichiometric aldol reactions provide general methods which overcome a number of the above limitations pointed out for catalytic aldol reactions. A common feature to stoichiometric methods is that no elimination occurs under the reaction conditions and thus aldols or ketols are obtained on work-up. Condensation products, whenever wanted, must be obtained in a separate dehydration step.Direct enolate aldol reactions
Ketones are moderate carbonacids and strong bases are required for their quantitative deprotonation. Although the variety of bases is larger in practice, lithium diisopropylamide (LDA) and sodium hydride may be taken here as commonly convenient reagents for the generation of enolates. Protic media are incompatible with these strong bases and tetrahydrofuran is the most frequent solvent. Absolute exclusion of moisture and oxygen are also fundamental experimental precautions. Reactions are carried out by deprotonation of the donor with an equivalent amount of base, and then addition of the acceptor. The immediate reaction adduct is an alkaline aldolate which, on neutralization with an equivalent amount of acid, affords the aldol or ketol (Scheme 8). As a consequence of the prior generation of the enolate, the aldolization is thermodynamically favoured and irreversible in practice: a feature that determines many of the differences found between catalytic and stoichiometric aldol reactions. Thus, ketones now react conveniently as acceptors. However, a significant limitation is met for aldehydes as donors, as their deprotonation is problematic, a feature which is usually circumvented by conversion of the aldehyde to an aldimine (see below). Further, the basicity of enolates may cause proton exchange prior to the addition, a difficulty in cross aldolizations, and constrains the use of reactants with groups sensitive to basic reagents. Exchange with transition metals, such as zirconium or titanium may circumvent this problem.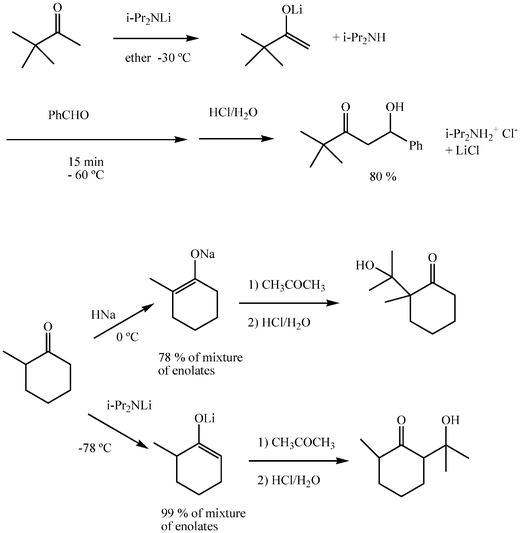 | ||
| Scheme 8 | ||
Regioselective deprotonation of asymmetric ketones that have similar acidity in their α and α′ positions is feasible by the appropriate choice of conditions. Thus, under conditions for kinetically controlled deprotonation (low temperature, short time, strong base in equivalent or excess amounts in the solution), the less substituted enolate predominates and under thermodynamically controlled conditions (near room temperature, adequate time for equilibration, limited amount of base in solution), the more substituted enolate is generated preferentially. (Scheme 8).
Aldol additions of generated lithium enolates are diastereoselective. The trend is determined by the geometry of the enolate and can be predicted with the aid of the Zimmerman–Traxler model, which is usually valid for lithium and boron enolates but not for other alkali metals or tetraalkylammonium ions as counterions. As a general rule, ethyl ketones with bulky carbonyl substituents afford the Z-enolate on deprotonation and this leads to syn ketols when added to aldehydes (Scheme 9). The synthesis of acyclic anti ketols is usually problematic, but for cyclic ketones no other enolate can result but the E-enolate and anti ketols are then obtained. Another effect of the aforementioned metal exchange is that it promotes higher syn selectivities.
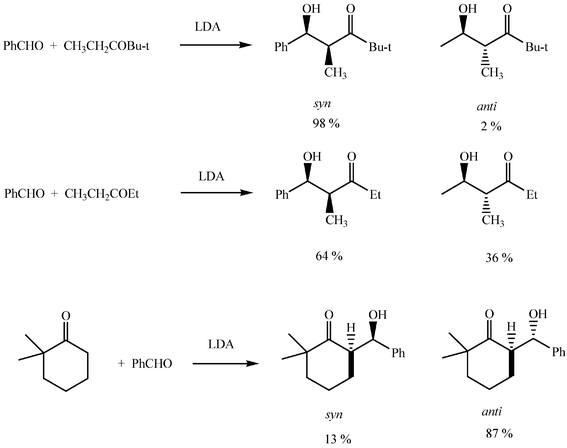 | ||
| Scheme 9 | ||
When attention is paid to safety and environmental issues associated with the aldol addition of stoichiometrically generated enolates, it is found first of all that bases for the quantitative generation of enolates are not bulk products. Although commercially available, tetrahydrofuran solutions of lithium amides are frequently prepared in situ from butyllithium and the corresponding amine. Sodium hydride is a commercial product that may ignite spontaneously in moist air and explodes in contact with water, DMSO or DMF (>25 °C). Ethereal solvents are highly flammable, form explosive peroxides, are moderately toxic by inhalation and there is some evidence of carcinogenic activity for THF.10 For an estimation of the atom economy it would not be unreasonable now to consider the generation of LDA from butyllithium, but restriction to the reagents actually used is preferred at the present level of study (Table 1). Waste comprises isopropylamine and lithium chloride for LDA as base and hydrogen and sodium chloride on use of sodium hydride. Transition metal exchange is certainly accompanied by a substantial increase of molar waste.
For the aldol reactions with aldehydes as donors, quantitative generation of the anions of their imines, usually their cyclohexylimine, with a lithium dialkylamide and addition to the acceptor aldehyde or ketone is achieved (Scheme 10). The resulting hydroxy imine is afterwards hydrolysed to the corresponding hydroxy aldehyde. This aldol reaction is known as the Wittig aldol reaction.
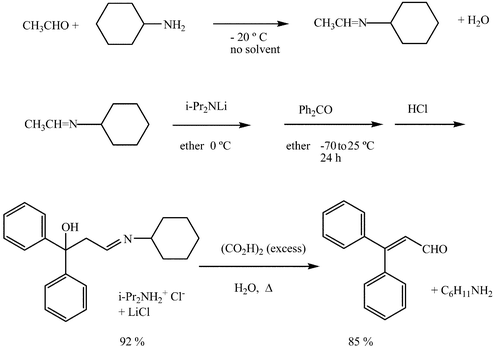 | ||
| Scheme 10 | ||
Cyclohexylamine is a severe human skin irritant, a suspected carcinogen and is included in the EPA “Extreme Hazardous Substances List”. Due to its high water solubility it is most probably discarded along with water effluents.10 Other waste products are the same as in the direct enolate reaction (Table 1).
Silyl enol ether aldol reactions
Silyl enol ethers as intermediates in the aldol reaction may improve regioselectivity and stereoselectivity of the enolate additions and offer the alternative possibility of carrying out the aldol addition under the Lewis acid promoted conditions known as the Mukaiyama reaction.13,14Silyl enol ethers are prepared by stoichiometric deprotonation of the ketone and silylation of the enolate, most frequently with trimethylchlorosilane. When the derivative of the less substituted enolate of asymmetric ketones is to be prepared, silylation of the enolate generated under kinetic conditions is carried out. However, deprotonation can also be promoted by a tertiary amine in the presence of the silylating agent when the thermodynamic silyl enol ether is sought (Scheme 11). The resulting silyl enol ethers obtained by one or other procedure can be resolved and purified by distillation or chromatography and thus the right regio- and stereo-isomers can be isolated.
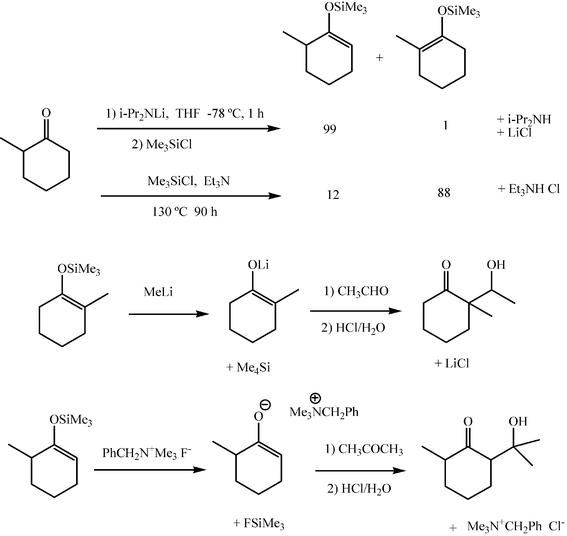 | ||
| Scheme 11 | ||
Once purified, silyl enol ethers can be used for the aldol addition to any acceptor by generating unequivocally the enolate in situ. This can be done either with methyllithium, or with fluoride ion. The behaviour of the enolate may differ according to the reagent used, as the counterion in the first case is a lithium ion, whereas in the second it is a tetraalkylammonium ion. When silyl enol ethers are employed, regioselectivities and stereoselectivities mentioned above for lithium enolates are improved, but stereoselectivities may differ in their trends when fluoride is used as nucleophile for generation of the enolate.
In regard to safety and environment protection, it may be added here that trimethylchlorosilane is a side product in the bulk production of the basic monomers of silicones. Trimethylchlorosilane is found in the list of very dangerous substances and is highly flammable (f.p. −27 °C), toxic, irritant and corroding in contact with skin, eyes and mucous membranes.10 Methyllithium in hexane (self ignition) and a variety of tetraalkylammonium fluorides are commercially available. As for concomitants, tetramethylsilane is very volatile (b.p. 26.6 °C) and most likely to be released to the atmosphere. Other waste products are usually discarded to water effluents (Table 1).
As referred to above, the silyl enol ether can react with an aldehyde or a ketone in the presence of a Lewis acid, most frequently titanium tetrachloride, in a nonbasic solvent, namely methylene dichloride or toluene, according to the named Mukaiyama aldol reaction (Scheme 12). The undesirable retro aldol process, present in the catalytic aldol reactions is inhibited here by formation of a stable titanium chelate, which is hydrolysed by water on work-up and excellent yields are thus obtained. Although high stereoselectivities are rarely obtained by the Mukaiyama aldol reaction, many recent asymmetric reactions are based on Lewis acid catalysed silyl enol ether aldol additions.
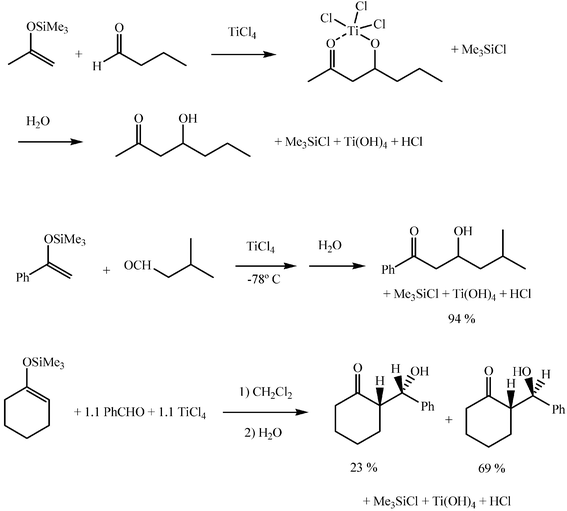 | ||
| Scheme 12 | ||
Titanium tetrachloride is an intermediate in the preparation of titanium dioxide and of titanium metal, resulting from chlorination of rutile. It is an irritant, corroding and toxic in contact with skin, eyes and mucous membranes and may ignite spontaneously in air.10 Molar waste for the Mukaiyama aldol reaction is very high (Table 1) and use of methylene dichloride as solvent is a further cause of concern.
Boron enolate aldol reactions
Boron enolates are generated by reaction of a ketone with dialkylboron trifluoromethyl sulfonates (triflates) or chlorides in the presence of a tertiary amine. Interestingly, they react with carbonylic compounds, without need for a catalyst, in methylene dichloride, THF or pentane as solvent. The resulting ketol boron ester is oxidatively hydrolysed with aqueous hydrogen peroxide (9–10 equivalents at pH 7) or with MoO5–pyridine–HMPA to afford the ketol.14The main advantage offered by the boron mediated aldol reaction is the improved stereocontrol usually achieved. The stereochemical outcome can be predicted through the same Zimmermann–Traxler model applied for lithium enolates, but now the stereoselectivity of the addition is superior. In accordance with that model, Z-boron enolates lead to syn (erythro) ketols, on addition to aldehydes, whereas the anti (threo) diastereoisomers are obtained from the E-boron enolates. The right choice of reagents allows the right geometry of the boron enolate to be attained. Thus, small ligands and a good leaving group on the boron and a bulky amine usually lead to the Z-boron enolate of simple ethyl ketones, whereas sterically demanding boron ligands, a less reactive leaving group and small amines usually promote the E-boron enolate (Scheme 13). A good control of the diastereoselectivity of the boron enolate aldol addition enables satisfactory enantioselectivities to be achieved by use of easily attainable chiral boron reagents. Enantioselective aldol additions can thus be obtained without an increase in the amount of waste associated with the use of chiral auxiliaries.
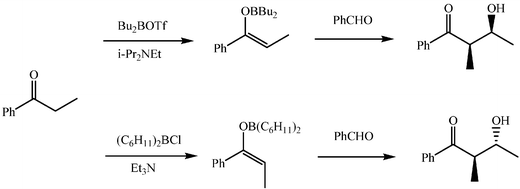 | ||
| Scheme 13 | ||
A number of dialkylboron triflates are commercially available in dichloromethane, hexane, or ether solution. Other triflates and the chlorides can be prepared from the dialkylboranes and trifluoromethanesulfonic acid. On the other hand, the starting dialkylboranes must be prepared beforehand by hydroboration. Boron enolates are most sensitive to Lewis acids and to moisture. They must be freshly prepared and their aldol reactions must be carried out under strict anhydrous conditions. No adequate toxicity record is available, but dibutylboron triflate is extremely flammable and dangerous in contact with the skin, according to a commercial catalogue. The actual figures for molar waste depend on the alkyl groups of the boron reagent and on the tertiary amine used (Table 1).
In conclusion, this section has shown that stoichiometric aldol reactions substantially improve the aldol reaction as a general synthetic method, especially in selectivities which cannot be attained by the general base or acid catalytic method. However this improvement is accompanied by a significant increase in molar waste and in hazards associated with toxicity, flammability or susceptibility to contribute to environmental pollution.
Recent and greener aldol reactions
Introduction
Some of the recent contributions to the green methodology of the aldol reaction, along with their advantages and limitations are introduced here. It should be said at the outset that these contributions may or may not be the result of green thinking, but in a number of instances they may open up routes for highly efficient, environmentally friendly C–C bond forming processes. Except for the aqueous Mukaiyama reaction, recently published methods are catalytic. This is promising in the context of methods meant to be green, but some of the constraints found above for the early catalytic aldol reactions are met again. Thus, many of these recent contributions can be applied only to a limited number of ketones as donors and aldehydes as acceptors. However, a few of the recent findings excel in the achievement of the control of the stereochemistry of aldolizations.Solvent free catalytic aldol reactions
Aldol reactions of small molecular weight aldehydes and ketones are carried out under catalytic conditions without any solvent. This is the case for acetaldehyde, butanal, acetone, or cyclohexanone. However, few recent reports of aldol condensations of higher molecular weight aldehydes or ketones under solvent-free conditions have been described. Thus, excellent condensation yields have been obtained by simple mixture of donor, acceptor and a molar equivalent of sodium hydroxide by grinding them together with a mortar and pestle or in a vibrating mill and a quite simple isolation of products (Scheme 14).15,16 Microwave activation has proved convenient for Claisen–Schmidt and related reactions carried out with or without solvent, either directly or by use of acetals.17–19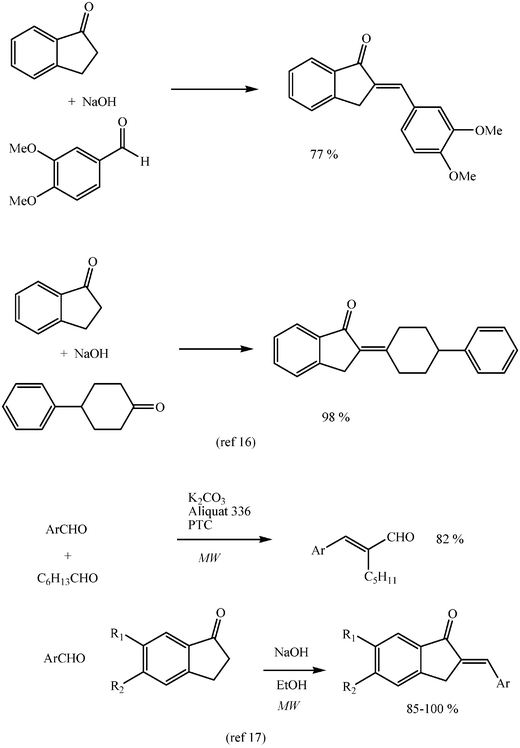 | ||
| Scheme 14 | ||
Catalytic aldol reactions in water
Aldol addition of a limited number of ketones to nitrobenzaldehydes has recently been reported to occur in water with a catalytic amount of sodium carbonate, to afford excellent yields of ketols.20 The microwave accelerating effect has also been shown for sodium hydroxide catalysed aldol condensations of aromatic aldehydes with acetone or acetophenone in aqueous medium.21Water at 250 °C (near-critical water, NCW) seems promising as a solvent for organic synthetic reactions, due to its solubility properties, comparable to those of polar organic solvents at room temperature and to its high dissociation constant. The latter property allows acid or base catalysed reactions to be carried out in NCW without need of added catalysts. Aldol reactions carried out under these conditions lead to rather low conversion yields (below 40%) of the condensation products. Oligomerization competes when alkyl aldehydes are used. On the other hand, butanone leads to regioisomeric mixtures.22 The Claisen–Schmidt condensation of benzaldehyde with acetone or acetophenone can be very selective and suitable for a large scale continuous process when carried out in water at 200 °C, especially because of the easy separation of products from the unreacted starting compounds.23
Reactions in supercritical fluids
No record has been found of aldol reactions in supercritical fluids, although a related asymmetric Mukaiyama reaction of the silyl enol ether of an ester of thioacetic acid in supercritical fluoroform has been described.24Reactions in ionic liquids
The self-condensation of propionaldehyde and the cross-condensation of the same aldehyde and 2-methylpentanal have been performed in ionic liquids. Best results with 1 M hydroxide containing butyldimethylimidazolinium hexafluorophosphate [bdmim][PF6] afford conversion yields comparable to those obtained in parallel condensations in water. Side products due to further aldol condensations of the first products are observed in the ionic liquids. However, significantly better selectivity results for the cross aldol condensation of propionaldehyde and 2-methylpentanal are obtained in [bdmim][BF4] than in water.25Reactions under heterogeneous catalysis
Heterogeneous catalysts for the aldol reaction offer the possibility of simpler separations of the catalyst from the reaction mixture. Though attractive due to their easy design, variety of porous system, hydrophobicity and hydrophilicity, zeolites have not proved good catalysts up to now for those aldol reactions susceptible to being useful in fine chemistry and pharmaceutical industries. Modified hydrotalcites and microporous silica seem more promising and are the object of much current work. Thus, a hydrotalcite modified with potassium t-butoxylate catalyses the condensation of acetone with a variety of substituted benzaldehydes very efficiently, with 100% conversions and isolated yields above 90% (Scheme 15).26,27 Although high yields are described for the aldol condensation of citral with excess butanone, regioselectivities found are still poor.28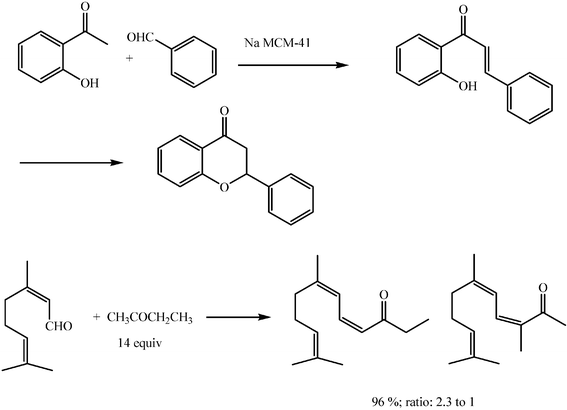 | ||
| Scheme 15 | ||
Synthesis on solid phase
Aldol reactions and related condensations have been carried out on solid insoluble supports. Either the donor or the acceptor may be linked to the support (Scheme 16). With all the constraints bound to solid supported reagents, this optional method may be convenient for some cross condensations. However, one problem encountered is related to the fact that in solid phase synthesis, reagents have to be used in excess. This can lead to side reactions. For instance, the enone that forms by reaction of a resin-bound aldehyde with a ketone may undergo Michael addition of the excess of ketone.29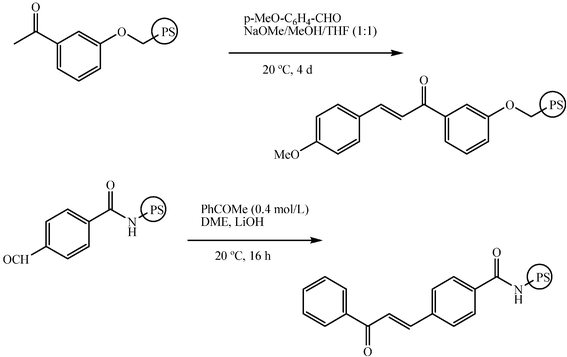 | ||
| Scheme 16 | ||
Synthetic chiral catalysts
The turn of the century has witnessed a strong interest for new synthetic chiral catalysts for aldol reactions.30–33 The general perspective of these asymmetric aldol reactions is beyond the scope of the present study and only two significant lines of work are mentioned here. Good yields and enantioselectivities for asymmetric aldol additions have been described by Trost and coworkers for aryl and alkyl aldehydes and excess of acetone, acetophenone or other aryl methyl ketones with a bimetallic chiral zinc catalyst.34,35 According to Shibasaki and coworkers, good results are obtained for a wider range of ketones with a lanthanum potassium bimetallic trisbinaphtoxide as catalyst,36 whereas α-hydroxyacetophenones react diastereoselectively and enantioselectively in the presence of a complex of diethylzinc and a linked BINOL (Scheme 17).37 Small amounts of catalyst are used (3% to 10%) in all these cases and it may be assumed that catalysts are easily recovered and reused. Although diethylzinc is consumed in any case in two of these procedures, the estimated molar waste is small (zinc chloride: 13.6 g mol−1) and thus, these reactions show a high atom economy, especially if the catalysts are recovered.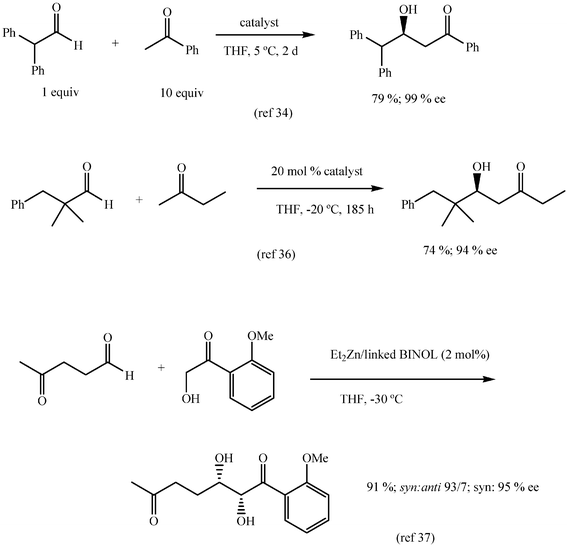 | ||
| Scheme 17 | ||
Biocatalysed aldol reactions
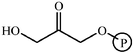
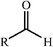
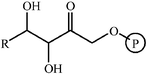
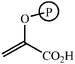
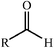
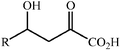
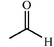
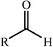
Aldolase enzymes and antibodies may catalyse aldol reactions.38 Aldolases are donor specific, diastereoselective and enantioselective, but not specific to the acceptor. They are limited thus to their specific donors, but a variety of aldehyde acceptors can be used. Plant aldolases do not require presence of metals in the active site and catalyse the aldol reaction through an imine (Schiff base) intermediate. Aldolases found in bacteria and fungi are usually Zn-dependent. Some typical donors are shown in Table 2. One of the relevant features of enzyme catalysed aldol additions is that the stereochemical outcome is entirely determined by the enzyme, and any possible enantiomer can be prepared by the right choice of the enzyme (Scheme 18).38 Synthesis of highly functionalised compounds is feasible through biocatalytic aldol reactions (Scheme 19). The specificity in the recognition of donors is certainly a serious limitation of biocatalytic procedures. For this reason it is especially interesting that mutant non-donor specific aldolases have been found by Wong (Scheme 20).39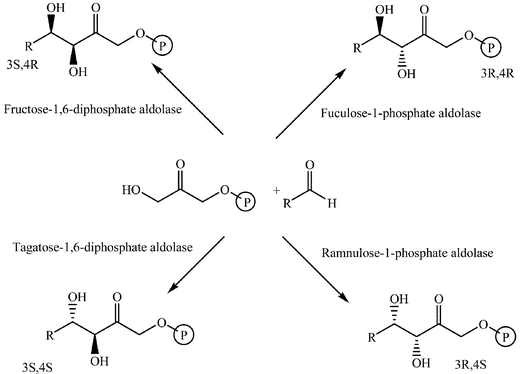 | ||
| Scheme 18 | ||
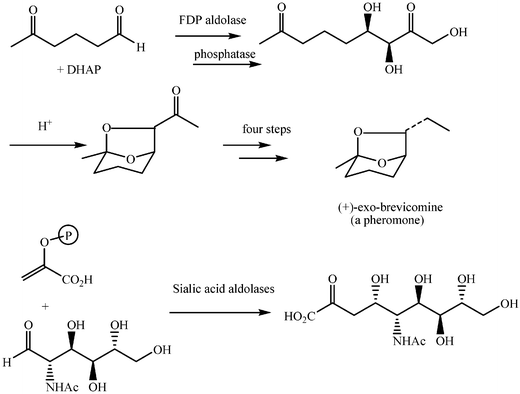 | ||
| Scheme 19 | ||
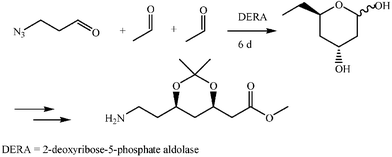 | ||
| Scheme 20 | ||
| Donor | Acceptor | Product |
|---|---|---|
| a Reference 38. | ||
|
|
|
|
|
|
|
|
|
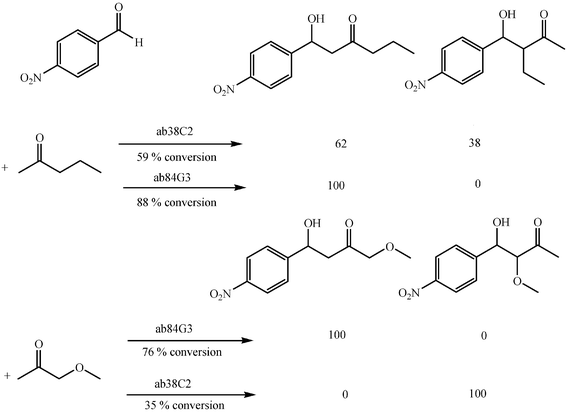
Recent findings by Gouverneur and coworkers show that the regioselectivity of the aldol addition of alkyl and hetero-substituted ketones to aldehydes can be reversed by the right choice of antibody as catalyst (Scheme 21).40
 | ||
| Scheme 21 | ||
Biomimetic catalyst aldol reactions
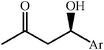
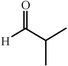
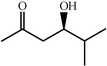
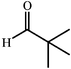
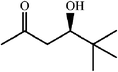
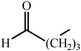
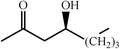
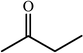
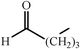
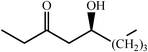
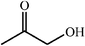
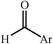
One of the most interesting recent contributions to the aldol reaction is the use of proline as catalyst. Intramolecular aldol condensations promoted by proline (with enantioselective dehydration) have been known since the early 1970s and referred to as the Hajos–Eder–Sauer–Wiechert reaction, an enantioselective Robinson annulation. However, catalysis of intermolecular aldol conversions by L-proline has been recognized only recently. The first report of excellent yields and enantioselectivities for additions of excess acetone to a limited number of aryl and α-branched alkyl aldehydes in DMSO at room temperature fostered a continuous stream of contributions, with results up to 2002 already reviewed,41–44 and the mechanism intensively studied.45 Some relevant results are presented in Tables 3 and 4. The significant point here is that proline raises the expectation of providing green procedures for aldol reactions and, what is especially interesting, combined with high diastereoselectivities and enantioselectivities. Although used in relatively high amounts as catalyst (20–25%), proline is inexpensive, easily available in both its R- and S-enantiomeric forms, essentially non-toxic, water soluble and easily separated from products for reuse while maintaining enantioselectivity and yield. Proline as catalyst can be regarded as a biomimetic of type I aldolase enzymes and antibodies, as the conversion occurs through the enamine of the donor and the stereochemical outcome is in agreement with a cyclic transition state similar to that depicted by the Zimmermann–Traxler model, where the carbonyl group of the acceptor aldehyde is proton bound to the carboxyl group of proline.41,42| Donor | Acceptor | Product | Yield (%) | dr (anti∶syn) |
ee (%) | Ref. |
|---|---|---|---|---|---|---|
| a Reports of aldolizations with yields below 50% have been omitted. b DMSO as solvent, unless otherwise stated. c Chloroform as solvent. | ||||||
|
|
|
62–94 | 60–85 | 41 | |
|
|
97 | 96 | 41 | ||
|
|
81 | 99 | 42 | ||
|
|
75 | 73 | 48 | ||
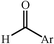
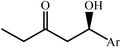
|
|
|
65 | 77 | 48 | |
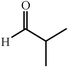
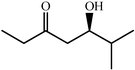
|
|
80 | 80 | 48 | ||
|
|
65 | 58 | 48 | ||
|
|
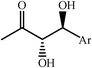
|
60–95 | 3∶2 to 20∶1 |
67–99 | 49 |
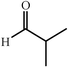
|
|
62 | 20∶1 |
99 | 49 | |
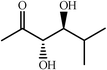
|
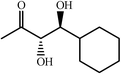
|
60 | 20∶1 |
99 | 49 | |
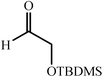
|
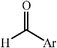
|
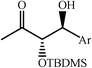
|
68–90 | 50 | ||
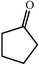 c
c
|
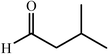
|
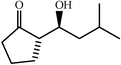
|
55 | 5∶2 |
20 | 42 |
|
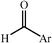
|
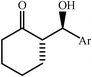
|
85 | 1∶1 |
85 | 42 |
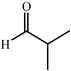
|
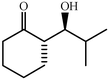
|
68 | 20∶1 |
97 | 43 | |
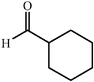
| Donor | Acceptor | Product | Yield (%) | dr (anti∶syn) |
ee (%) | Ref. |
|---|---|---|---|---|---|---|
| a Reports of aldolizations with yields below 50% have been omitted. b DMF as solvent, unless otherwise stated. c DMSO as solvent. | ||||||
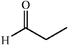
|
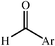
|
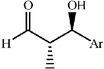
|
81 | 3∶1 |
99 | 46 |
|
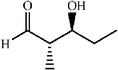
|
80 | 4∶1 |
99 | 46 | |
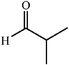
|
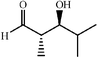
|
82 | 24∶1 |
99 | 46 | |
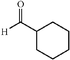
|
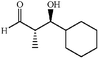
|
87 | 14∶1 |
99 | 46 | |
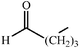 c
c
|
|
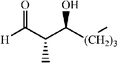
65 | 58 | 48 | ||
|
|
84 | 5∶1 |
99 | 52 | |
|
|
88 | 3∶1 |
97 | 46 | |
|
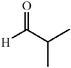
|
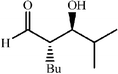
|
80 | 24∶1 |
98 | 46 |
|
|
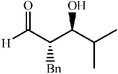
|
75 | 19∶1 |
91 | 46 |
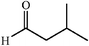
|
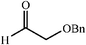
|
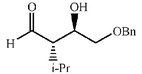
|
64 | 4∶1 |
94 | 52 |
|
|
|
73 | 4∶1 |
98 | 52 |
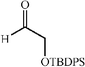
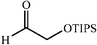
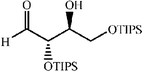
|
 c
c
|
|
92 | 4∶1 |
95 | 52 |
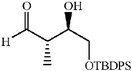
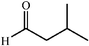
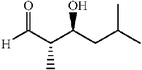
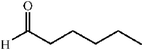
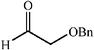
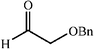
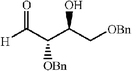
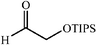
The scope of the catalysis by proline has been expanded steadily and the addition to α-unbranched aldehydes has been successfully carried out in acetone or acetone–chloroform.42 Cross reaction of aldehydes has also been achieved.46,47 Butanone reacts conveniently through its methyl group, cyclohexanone and cyclopentanone have also been successfully used, but larger ketones, namely 3-pentanone or acetophenone do not react.42,48 Interestingly, proline promoted aldol additions of free or protected hydroxyacetone as donor occur with excellent regioselectivity, diastereoselectivity and enantioselectivity to give α,β-dihydroxy ketones.42,49,50 The potential of proline as catalyst is shown by the trimerization of propionaldehyde to give cyclic hemiacetals in 53% yield as anomeric mixtures of two diasteroisomers (Scheme 22),51 or by the self-aldolization of alkyl or silyl protected hydroxyacetaldehyde to give erythrose, as a first step for a simple synthesis of hexoses (see Table 4).52
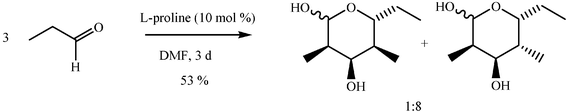 | ||
| Scheme 22 | ||
Not surprisingly, the outcome of the first results fostered much interest in finding related biomimetic catalysts that could overcome some of the limitations of proline. Thus, pyrrolidine and acetic acid jointly catalyse the cross aldol reaction of α-branched alkyl aldehydes as donors, when proline is not effective, to give quaternary carbon aldols of otherwise difficult access.53 Several acyclic and cyclic amino acids and their derivatives have been assayed and 5,5-dimethylthiazolidinium-4-carboxylic acid compares well with proline.48 Other modifications involve some synthetic peptides that have been prepared and evaluated as catalysts.54 The amide resulting from proline and enantiomerically pure 1,2-diphenylethanolamine has proved very efficient in the aldol addition of acetone to cyclohexylcarboxaldehyde (Scheme 23) and is in line with a new strategy of catalysis in which a carbonyl group is activated by a double proton bond.55,56
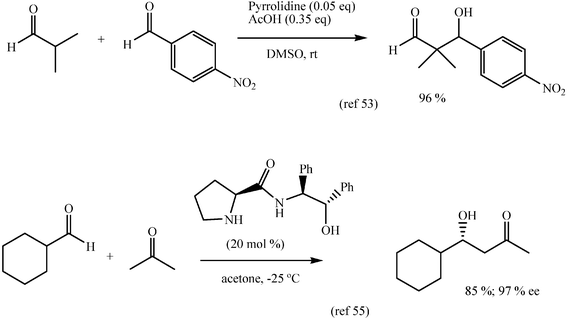 | ||
| Scheme 23 | ||
One of the drawbacks of the first proline catalysed aldol reactions is the need for aprotic solvents, namely DMSO, DMF or chloroform. The possibility of carrying out the reaction in water must then be taken as an interesting improvement and it has been reported that acetone adds to p-nitrobenzaldehyde in excellent yield in a phosphate buffered aqueous solution containing sodium dodecyl sulfate (SDS) in the presence of proline. Water inverts the regiochemical trend found for the addition of butanone in DMSO and the product resulting from attack by the methylene group predominates. However, aldol additions in water are poorly stereoselective and proline can be substituted on some occasions by pyrrolidine, by (S)-1-(2-pyrrolydinylmethyl)pyrrolidine, or by nornicotine.57–60 A simple and interesting modification of proline, that allows its use in water, consists of the Zn–proline complex, which is easily prepared from zinc acetate and two equivalents of proline. This complex is water soluble and water stable. Addition of excess acetone to p-nitrobenzaldehyde in water affords excellent yields, though only moderate enantioselectivities are observed. Interestingly, zinc complexes of other amino acids, namely lysine and arginine are also efficient catalysts.60
Other solvents have been studied with the common purpose of avoiding DMSO or DMF. Thus, a polyfluorous proline derivative has also been prepared and used as catalyst in benzene trifluoride solvent, with results similar to those obtained with proline in DMSO, although with a much simpler isolation procedure, according to the authors.61 Ionic liquids have proved convenient media as solvents for proline promoted aldol reactions, with good enantioselectivities being obtained, but with no remarkable advantages over DMSO or DMF.62,63 As another form of avoiding use of DMSO, the reaction of acetone and several aldehydes has been carried out under pressure, with the ketone as solvent. Good yields of the corresponding ketols are obtained for benzaldehydes, but enantioselectivities seem to decrease at high pressure.64
Immobilization of proline for easier separation of products has been carried out by linkage of proline through the carboxyl group to a polystyrene resin. The resin thus functionalised promotes enantioselective aldolizations and can be reused with no loss of activity.65 Grafting in mesoporous MCM-41 gives poorer yields and enantioselectivities.66
Silyl enol ether aldol reactions in water
Aldol reactions of silyl enol ethers promoted by typical Lewis acids must be carried out under strict anhydrous conditions. However, the development of Lewis acids tolerant to water has allowed successful stereoselective aldol reactions of silyl enol ethers in water. Certain metal trifluoromethylsulfonates (triflates) are water soluble and water tolerant, air-stable and easy to handle, such as Pb(OTf)2, La(OTf)3, Ce(OTf)3, or Ga(OTf)3, and others. For instance, a 20 mol% solution of gallium triflate with a semi-crown ligand promotes the Mukaiyama reaction of the silyl enol ether of propiophenone with aryl and alkyl aldehydes in a water–ethanol (9∶1) solvent system to give good to excellent yields of the ketols and promising diastereoselectivity (syn∶anti 80∶20) and enantioselectivity (ee for syn ketol 80%).67,68
Moderate to good yields of aldol adducts can also be obtained in water by use of silyl enol ethers in the presence of a surfactant copper salt, namely copper dodecylsulfate, and a carboxylic acid. The addition is diastereoselective and, in the presence of a chiral bis-oxazoline, enantioselective (Scheme 24).69
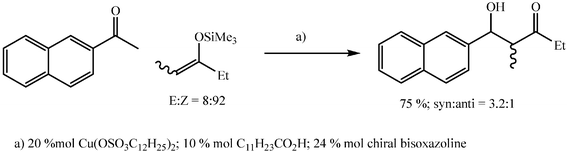 | ||
| Scheme 24 | ||
Another interesting catalyst for water aldol reactions of silyl enol ethers is diphenylborinic acid, which in the presence of benzoic acid and SDS promotes additions to aldehydes to give syn-substituted β-hydroxy ketones in high diastereoisomeric excesses.68
Catalytic aldol reactions of α,β-unsaturated ketones under reduction conditions
Recent promising findings by Krische and coworkers show that in situ catalytic generation of metallic enolates occurs when α,β-unsaturated ketones or aldehydes are subject to reduction by molecular hydrogen in the presence of a cationic rhodium catalyst capable of causing heterolytic activation of molecular hydrogen. The enolates thus generated add efficiently to the carbonyl group of aldehydes or ketones present in the reaction medium, affording the corresponding aldols or ketols. With some constraints, the procedure can be conveniently applied to both inter- and intra-molecular conversions (Scheme 25).70–73 The significance of the reaction resides in its ability to ensure a regioselective course in the preparation of aldols and ketols of difficult access when starting from simple aldehydes or ketones and using basic or acid catalysis. Further development is expected to expand the scope of the procedure and to achieve both diastereoselectivity and enantioselectivity.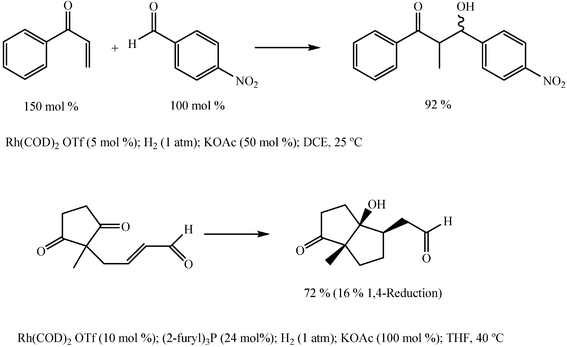 | ||
| Scheme 25 | ||
Catalytic aldol reactions of allylic alcohols
Motherwell and coworkers described in 1999 that rhodium catalysed isomerization of allylic lithium alkoxides allowed regioselective generation of the metal enolates of ketones or aldehydes and that these enolates added as expected to aldehydes and ketones in an aldol fashion.74 However, although allowing regioselective preparation of aldols and ketols, the method needed a waste producing stoichiometric deprotonation of allylic alcohols with butyllithium. Fortunately, Greé and coworkers found that the aldol process can occur satisfactorily with metal enolates catalytically generated directly from allylic alcohols with iron pentacarbonyl or better with rhodium or ruthenium complexes.75–77Later on Li and coworkers found that the reaction could conveniently be carried out in water and in ionic liquids with ruthenium dichloride triphenylphosphine complex (Scheme 26).78,79
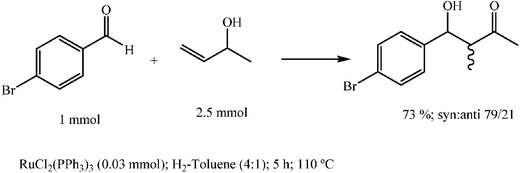 | ||
| Scheme 26 | ||
Conclusion
It has been shown that the general catalytic aldol reaction can be regarded as substantially waste free and safe, but subject to a number of limitations as a method of broad general preparative application. Recently developed catalytic systems have added the achievement of good to excellent stereoselectivities. Stoichiometric procedures expand the scope of the aldol reaction as a general synthetic method and offer the possibility of directing their selectivity trends but at the price of a significant increase in both molar waste and hazard. The recent introduction of water-compatible Lewis acids for the Mukaiyama aldol reaction partly overcomes the safety and environmental drawbacks of silyl enol ether based stoichiometric procedures.Viewing the present scene, consideration of the sustainability of the reaction would indicate application of catalytic procedures whenever possible, before going to stoichiometric procedures and the direct use of preformed enolates before attempting silyl or boron enol ether mediated stoichiometric reactions.
Biocatalysis and biomimetic catalysis, though not exclusively, seem at present the most promising lines to follow for the development of green aldol reactions. Compatibility of biomimetic catalysts with safe protic solvents or with systems from which products can be easily separated whilst retaining the catalyst would be welcome, especially if conversion yields and selectivities are maintained or improved. However, the search for clean and safe methods in keeping with the achievement of the stoichiometric reactions should also be fostered.
References
- M. B. Smith and J. March, Advanced Organic Chemistry, Wiley-Interscience, New York, 5th edn., 2001, p. 1218 Search PubMed; F. A. Carey and R. J. Sundberg, Advanced Organic Chemistry Part B, Kluwer Academic/Plenum Press, New York, 4th edn., 2000, p. 57 Search PubMed; H. O. House, Modern Synthetic Reactions, Benjamin, Menlo Park, CA, 2nd edn., 1972, p. 629 Search PubMed; J. H. Furhopp and G. Li, Organic Synthesis, Wiley-VCH, Weinheim, 3rd edn., 2003, p. 44 Search PubMed.
- Houben–Weyl, Die Methoden der Organische Chemie, Georg Thieme Verlag, Stuttgart, 1954, Band VII, Teil 1, p. 76 Search PubMed.
- A. T. Nielsen and W. J. Houlihan, Organic Reactions, John Wiley, New York, 1968, vol. 16, p. 1 Search PubMed.
- H. B. Mekelburger and C. S. Wilcox, in Comprehensive Organic Synthesis, ed. B. M. Trost and I. Fleming, Pergamon Press, 1991, vol. 2, p. 99 Search PubMed; C. H. Heathcock, in Comprehensive Organic Synthesis, ed. B. M. Trost and I. Fleming, Pergamon Press, 1991, vol. 2, p. 133 Search PubMed; C. H. Heathcock, in Comprehensive Organic Synthesis, ed. B. M. Trost and I. Fleming, Pergamon Press, Oxford, UK, 1991, vol. 2, p. 181 Search PubMed.
- M. M. Green and H. A. Wittcoff, Organic Chemistry Principles and Industrial Practice, Wiley-VCH, Weinheim, 2003 Search PubMed.
- P. T. Anastas and J. C. Warner, Green Chemistry: Theory and Practice, Oxford University Press, Oxford, 1998, p. 29 Search PubMed; N. Winterton, Green Chem., 2001, 3, G73 Search PubMed.
- A. D. Curzons, D. J. C. Constable, D. N. Mortimer and V. L. Cuningham, Green Chem., 2001, 3, 1 RSC.
- M. Eissen and J. O. Metzger, Chem.—Eur. J., 2002, 3581–3585; M. Eissen, J. O. Metzger, E. Schmidt and U. Schneidewind, Angew. Chem., Int. Ed., 2002, 41, 414 CrossRef CAS; M. Eissen, R. Mazur, H. G. Quebbemann and K. H. Pennemann, Helv. Chim. Acta, 2004, 87, 524 CrossRef CAS.
- M. Lancaster, Green Chemistry, RSC, Cambridge, 2002, p. 69 Search PubMed; M. Lancaster, in Handbook of Green Chemistry and Technology, ed. J. Clark and D. Macquarrie, Blackwell, Oxford, 2002, p. 10 Search PubMed.
- R. J. Lewis, Sr., Hazardous Chemical Desk Reference, Wiley-Interscience, New York, 5th edn., 2002 Search PubMed.
- J. P. Guthrie, J. Am. Chem. Soc., 1991, 113, 7249 CrossRef CAS.
- J. B. Conant and N. Tuttle, in Organic Syntheses Coll. Vol. 1, John Wiley, New York, 1941, p. 199 Search PubMed.
- T. Mukaiyama, Organic Reactions, John Wiley, New York, 1982, vol. 28, p. 237 Search PubMed; T. H. Chan, in Comprehensive Organic Synthesis, ed. B. M. Trost and I. Fleming, Pergamon Press, Oxford, UK, 1991, vol. 2, p. 595 Search PubMed.
- D. A. Evans, J. V. Nelson, E. Vogel and T. R. Taber, J. Am. Chem. Soc., 1981, 103, 3099 CrossRef CAS; C. J. Cowden and I. Paterson, Organic Reactions, John Wiley, New York, 1997, vol. 51, p. 1 Search PubMed; Houben–Weyl, Methods of Organic Chemistry, Georg Thieme Verlag, Stuttgart, 1995, vol. E 21b, p. 1603 Search PubMed.
- K. Tanaka, Solvent-free Organic Synthesis, Wiley-VCH, Weinheim, 2003, p. 47 Search PubMed.
- C. L. Raston and J. L. Scott, Green Chem., 2000, 2, 49 RSC.
- P. Lidström, J. Tierney, B. Wathey and J. Westman, Tetrahedron, 2001, 57, 9225 CrossRef CAS.
- J. S. Yadav, B. V. S. Reddy, A. Nagaraju and J. A. R. P. Sarma, Synth. Commun., 2002, 32, 893 CrossRef CAS.
- D. F. Huang, J. X. Wang, Y. Hu, Y. Zhang and J. Tang, Synth. Commun., 2002, 32, 971 CrossRef CAS.
- G. W. Wang and Y. W. Dong, Org. Process Res. Dev., 2004, 8, 18 Search PubMed.
- G. L. Kad, K. P. Kaur, V. Singh and J. Singh, Synth. Commun., 1999, 29, 2583 CAS.
- S. A. Nolen, C. L. Liotta, C. A. Eckert and R. Glaser, Green Chem., 2003, 5, 663 RSC.
- C. M. Comisar and P. E. Savage, Green Chem., 2004, 6, 227 RSC.
- D. Prajapati and M. Gohain, Tetrahedron, 2004, 60, 815 CrossRef CAS.
- C. P. Mehnert, N. C. Dispenziere and R. A. Cook, Chem. Commun., 2002, 1610–1611 RSC.
- F. Figueras and J. Lopez, in Fine Chemicals through Heterogeneous Catalysis, ed. R. A. Sheldon and H. van Bekkum, Wiley-VCH, Weinheim, 2001, p. 327 Search PubMed.
- M. J. Climent, A. Corma, S. Iborra and A. Velty, J. Catal., 2004, 221, 474 CrossRef CAS.
- M. J. Climent, A. Corma, S. Iborra and A. Velty, Green Chem., 2002, 4, 474 RSC.
- F. Z. Dörwald, Organic Synthesis on Solid Phase, Wiley-VCH, Weinheim, 2nd edn., 2002, p. 180 Search PubMed.
- T. D. Machajewski and C. H. Wong, Angew. Chem., Int. Ed., 2000, 39, 1352 CrossRef CAS.
- C. Palomo, M. Oiarbide and J. M. García, Chem.—Eur. J., 2002, 8, 36–44 CrossRef CAS.
- B. Alcaide and P. Almendros, Eur. J. Org. Chem., 2002, 1595 CrossRef CAS.
- B. Alcaide and P. Almendros, Angew. Chem., Int. Ed., 2003, 42, 858 CrossRef CAS.
- B. M. Trost and H. Ito, J. Am. Chem. Soc., 2000, 122, 12003 CrossRef CAS.
- B. M. Trost, E. R. Silcoff and H. Ito, Org. Lett., 2001, 3, 2497 CrossRef.
- N. Yoshikawa, Y. M. A. Yamada, J. Das, H. Sasai and M. Shibasaki, J. Am. Chem. Soc., 1999, 121, 4168 CrossRef CAS.
- N. Komagai, S. Matsunaga, T. Kinoshita, S. Harada, S. Okada, S. Sakamoto, S. K. Yamaguchi and M. Shibasaki, J. Am. Chem. Soc., 2003, 125, 2169 CrossRef CAS.
- K. Faber, Biotransformations in Organic Chemistry, Springer–Verlag, Berlin, 4th edn., 2000, p. 273 Search PubMed.
- J. Liu, C. C. Hsu and C. H. Wong, Tetrahedron Lett., 2004, 45, 2439 CrossRef CAS.
- V. Maggiotti, S. Bahmanyar, M. Reiter, M. Resmini, K. N. Houk and V. Gouverneur, Tetrahedron, 2004, 60, 619 CrossRef CAS.
- B. List, R. Lerner and C. F. Barbas III, J. Am. Chem. Soc., 2000, 122, 2395 CrossRef CAS.
- B. List, P. Pojarliev and C. Castello, Org. Lett., 2001, 3, 573 CrossRef CAS.
- B. List, Tetrahedron, 2002, 58, 5573 CrossRef CAS.
- E. R. Jarvo and S. J. Miller, Tetrahedron, 2002, 58, 2481 CrossRef.
- M. Arnó and L. R. Domingo, Theor. Chem. Acc., 2002, 108, 232 Search PubMed and references therein.
- A. B. Northrup and D. W. C. MacMillan, J. Am. Chem. Soc., 2002, 124, 6798 CrossRef CAS.
- P. M. Pihko and A. Erkkilä, Tetrahedron Lett., 2003, 44, 7607 CrossRef CAS.
- K. Sakhtivel, W. Notz, T. Bui and C. F. Barbas III, J. Am. Chem. Soc., 2001, 123, 5260 CrossRef CAS.
- W. Notz and B. List, J. Am. Chem. Soc., 2000, 122, 7386 CrossRef CAS.
- H. Liu, L. Peng, T. Zhang and Y. Li, New J. Chem., 2003, 27, 1159 RSC.
- N. D. Chowdary, D. B. Ramachary, A. Córdova and C. F. Barbas III, Tetrahedron Lett., 2002, 43, 9591 CrossRef CAS.
- A. B. Northrup, I. K. Mangion, F. Hettche and D. W. C. MacMillan, Angew. Chem., Int. Ed., 2004, 43, 2152 CrossRef CAS.
- N. Mase, F. Tanaka and C. F. Barbas III, Org. Lett., 2003, 5, 4369 CrossRef CAS.
- J. Kofoed, J. Nielsen and J. L. Reymond, Bioorg. Med. Chem. Lett., 2003, 13, 2445 CrossRef CAS.
- Z. Tang, F. Jiang, L. T. Yu, X. Cui, L. Z. Gong, A. Q. Mi, Y. Z. Jiang and Y. D. Wu, J. Am. Chem. Soc., 2003, 125, 5262–5263 CrossRef CAS.
- P. M. Pihko, Angew. Chem., Int. Ed., 2004, 43, 2062 CrossRef CAS.
- A. Córdova, W. Notz and C. F. Barbas III, Chem. Commun., 2002, 3024 RSC.
- Y. Y. Peng, Q. P. Ding, Z. Li, P. G. Wang and J. P. Cheng, Tetrahedron Lett., 2003, 44, 3871 CrossRef CAS.
- T. J. Dickerson and K. D. Janda, J. Am. Chem. Soc., 2002, 124, 3220 CrossRef CAS.
- T. Darbre and M. Machuqueiro, Chem. Commun., 2003, 1090 RSC.
- F. Fache and O. Piva, Tetrahedron: Asymmetry, 2003, 14, 139 CrossRef CAS.
- T. P. Loh, L. C. Feng, H. Y. Yang and J. Y. Yang, Tetrahedron Lett., 2002, 43, 8741 CrossRef CAS.
- P. Kotrusz, I. Kmentová, B. Gotov, S. Toma and E. Solcániová, Chem. Commun., 2002, 2510 RSC.
- Y. Segikuchi, A. Sasaoka, A. Shimomoto, S. Fujioka and H. Kutsuki, Synlett, 2003, 1655.
- G. Szollosi, G. London, L. Balaspiri, C. Somlai and M. Bartok, Chirality, 2003, 15, suppl. S, S90.
- D. Dhar, I. Beadham and S. Chandrasekaran, Proc. Indian Acad. Sci., Chem. Sci., 2003, 115, 365 Search PubMed.
- H. J. Li, H. Y. Tian, Y. J. Chen, D. Wang and C. J. Li, Chem. Commun., 2002, 2994 RSC.
- U. M. Lindström, Chem. Rev., 2002, 102, 2751 CrossRef.
- S. Kobayashi, Y. Mori, S. Nagayama and K. Manabe, Green Chem., 1999, 1, 175 RSC.
- H. Y. Jang, R. R. Huddleston and M. J. Krische, J. Am. Chem. Soc., 2002, 124, 15156 CrossRef CAS.
- R. R. Huddleston and M. J. Krische, Org. Lett., 2003, 5, 1143 CrossRef CAS.
- G. A. Marriner, S. A. Garner, H. Y. Jang and M. J. Krische, J. Org. Chem., 2004, 69, 1380 CrossRef CAS.
- P. K. Koech and M. J. Krische, Org. Lett., 2004, 6, 691 CrossRef CAS.
- L. J. Gazzard, W. B. Motherwell and D. A. Sandham, J. Chem. Soc., Perkin Trans. 1, 1999, 979 RSC.
- C. Crévisy, M. Wietrich, V. Le Boulaire, R. Uma and R. Greé, Tetrahedron Lett., 2001, 42, 395 CrossRef CAS.
- R. Uma, M. Davies, C. Crévisy and R. Greé, Tetrahedron Lett., 2001, 42, 3069 CrossRef CAS.
- C. Crévisy and R. Greé, J. Org. Chem., 2003, 68, 6392 CrossRef.
- M. Wang and C. J. Li, Tetrahedron Lett., 2002, 43, 3589 CrossRef CAS.
- X. F. Yang, M. Wang, R. S. Varma and C. J. Li, Org. Lett., 2003, 5, 657 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2004 |