Airborne sulfur and nitrogen in Finland—trends and exposure in relation to air transport sector
Tuija Ruoho-Airola*, Pia Anttila and Timo Salmi
Finnish Meteorological Institute, Air Quality, PO Box 503, 00101 Helsinki, Finland. E-mail: tuija.ruoho-airola@fmi.fi
First published on 20th November 2003
Abstract
We present the concentration trends and the atmospheric exposure of sulfur and nitrogen compounds in different transport sectors during the period 1981–2000, based on the air quality data of the Finnish Meteorological Institute. Sulfur and nitrogen concentrations in air and precipitation were assessed from background stations covering the whole country. A significant decrease of more than 60% was observed in the concentrations of all sulfur compounds throughout the country during 1981–2000. In the 1990's, significant trends were detected in all sulfur time series, with the exception of sulfate in precipitation at the northernmost stations. On the other hand, the concentrations of oxidized nitrogen compounds in air and precipitation have declined only slightly or not at all, especially in the 1990's. This is attributed to the fairly small domestic and European NOx emission reductions after 1981. The ammonium concentration in precipitation has declined by more than 50% during 1981–2000. The atmospheric sulfur exposure was found to be dominated by transport from the West and South-West sectors during summer, whereas in winter all the southward sectors were important. The largest decline in the sectoral sulfur concentrations has taken place already before 1991, with a more pronounced reduction during winter than during summer. The atmospheric nitrogen exposure was mainly dominated by transport from the sectors between west and south, but in the latter half of the study period there seems to have been a slight shift towards increased transport from the eastern and northern sectors.
1. Introduction
Acid deposition caused by the emissions of sulfur and nitrogen has been a major environmental problem in Finland and elsewhere in Scandinavia for decades.1,2 In the atmosphere, the primary emissions undergo reactions and are simultaneously diluted by diffusion and transported with air masses. Subsequent deposition can thus occur thousands of kilometres away from the emission sources, which makes acid deposition an international problem. The common need to reduce emissions led to the European wide UNECE LRTAP Convention (United Nations Economic Commission for Europe Convention on Long-Range Transport of Air Pollutants). Under the Convention, specific protocols aim to control the emissions to a level where the ecosystems could recover.Nitrate, sulfate and ammonium are major components of the atmospheric aerosol.3 While sulfate and ammonium are mainly found in the fine particle fraction, nitrates may occur in both the fine and coarse particle fractions. Once emitted in the atmosphere, gaseous ammonia (NH3) rapidly combines with the sulfate ion (SO42−) to form ammonium sulfate, (NH4)2SO4. Excess ammonia may then react with nitric acid to form ammonium nitrate, NH4NO3. Nitrate occurring in the coarse particle fraction is the result of reactions of nitric acid with sodium chloride or aerosol crustal material.4 In Finland, the nitrate to nitric acid molar ratio may vary at least between 0.25 and 2.5.5 Ammonium to ammonia molar ratios between 0.2 and 3 have been measured in southern Finland.5
In Finland, downward trends of the atmospheric sulfur and nitrogen compounds have previously been reported2,6–8 based on data measured at a limited number of stations within the EMEP (Co-operative Programme for Monitoring and Evaluation of the Long-Range transmission of Air Pollutants in Europe) and ICPIM (International Co-operative Programme Integrated Monitoring) monitoring programmes, both under the UNECE LRTAP Convention. The calculations in these studies extend up to 1998. Kulmala et al.9 have extensively investigated the air quality trends at the Finnish background stations for a time period ending in 1996. Sulfur in air and precipitation at all stations and the nitrogen compounds in precipitation at several stations were found to have significant downward trends–especially strong trends were obtained for the components connected with sulfur emissions. In the neighbouring Scandinavian countries, similar downward trends have been reported for the acidifying atmospheric compounds.10,11 In this paper, we continue the air quality trend calculations for the 16 background stations of the Finnish Meteorological Institute (FMI) to cover the years 1981–2000.
The concentrations of sulfur and nitrogen compounds in Finland are highly affected by long-range transport. Since the emissions in Europe have declined at a different pace in different countries, the origin of the air masses and the frequency of transport sectors have strongly influenced the mean concentration and the trends. This paper presents the first observation-based evaluation of the dependence of the atmospheric sulfur and nitrogen concentrations and the accumulated time-weighted exposure to these pollutants in different air transport sectors in Finland and combines the trends with the European emission reductions. We have investigated the distribution and change in time of exposure in eight transport sectors using daily monitoring values for the whole 15-year period 1986–2000. The classification of the monitoring data into different transport sectors is based on the 925 hPa trajectories provided by the EMEP/MSC-W (Meteorological Synthesizing Centre–West).
2. Materials and methods
2.1. Stations and monitoring
The trends of atmospheric sulfur and nitrogen compounds were studied using data from measurements performed at the background air quality stations of the Finnish Meteorological Institute. The stations are located in remote areas without nearby emission sources. With time, some station locations have been moved slightly due to changes in the surroundings of the stations, e.g. increased traffic. Fig. 1 shows the present location of the stations used in this study. The stations are grouped to represent different areas of the country when the results are discussed. The following abbreviations of the areas are later used in the text: SW southwestern Finland, SE southeastern Finland, C central Finland, SL southern Lapland, and NL northern Lapland.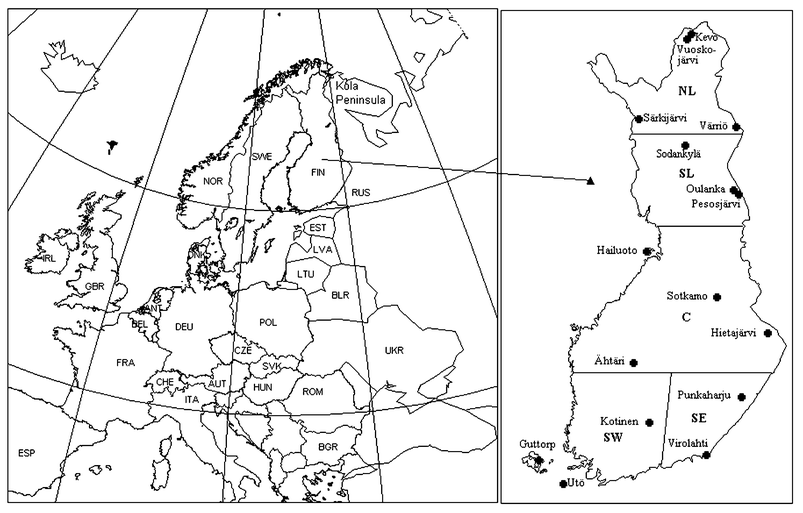 | ||
| Fig. 1 Map of the background air quality measuring stations of the Finnish Meteorological Institute. Different geographical areas are shown with bold letters: SW south-western Finland, SE south-eastern Finland, C central Finland, SL south Lapland, NL north Lapland. | ||
Four of the stations are located in a marine environment. The stations Utö (SW), Guttorp (SW) and Hailuoto (C) are located on Baltic Sea islands, while Virolahti (SE) is on the coast of the Gulf of Finland. Utö is situated on a rocky outer island, the surroundings of the other marine stations are sparsely populated agricultural areas with forests and fields. All other stations are inland stations, mostly surrounded by forest.
In the beginning of the study period gaseous SO2 was sampled by the H2O2 absorption method at all stations. Since 1989 an open-face filter pack with a NaOH impregnated Whatman 40 filter has been used at the EMEP stations. Particulate SO42− was sampled on a Whatman 40 filter installed in front of the SO2 sampling. The sum of the gaseous HNO3 and particulate NO3− was sampled by the same filter pack as the sulfur compounds. Sampling artifacts due to the evaporation of fine particulate nitrate from the first filter and recollection on the impregnated filter4 make the separation of these compounds by a simple aerosol filter pack unreliable. For the sum of the gaseous NH3 and particulate NH4+, a second filter line was used with a (COOH)2 impregnated Whatman 40 filter. For the deposition samples, the stations were equipped with NILU-type bulk deposition samplers. The analysis was performed at the Air Chemistry Laboratory of the Finnish Meteorological Institute (EN ISO/IEC 17025 accredited). At the end of the study period, all compounds were analysed by ion chromatography, the particles and gases after H2O extraction. In the beginning of the monitoring other methods were used: X-ray fluorescence for atmospheric SO42− (until 1987), the spectrophotometric Thorin method for SO42− in precipitation (until 1984) and SO2 (until 1989), and the indophenol blue method for the ammonium compounds (until 1992 in precipitation and 1995 in air). The NO2 concentration was monitored by the automated Saltzman method and a chemiluminescence monitor (since 1996). The sampling frequencies used for the components at different stations vary substantially: from continuous, in the case of NO2, to daily at the EMEP stations, and from weekly to monthly at the other stations.
The sampling and the analysis of the samples in the laboratory as well as the quality control of the whole chain were carried out using the best possible practices according to the guidelines given by the EMEP CCC12 (Chemical Co-ordinating Centre) and ICP IM.13 When a sampling or analysis method was changed, parallel sampling was performed in order to estimate the possible change in results. Thus the results of the SO2 concentration at the EMEP stations during 1981–1989 have been corrected for the overestimate of 1 µg m−3 of the older method.9 More detailed information on the background air quality measuring programmes and the methods is available from the EMEP CCC12 and in the annual report of the FMI air quality measurements.14
The accuracy, precision and detection limit of the monitoring data is controlled through the whole chain of measurements by means of field blanks, control samples and split samples. The EMEP CCC routinely tests the data quality by intercomparisons, which are performed annually for the chemical analysis and every now and then for the sampling methods. According to the EMEP CCC quality assessment,15 the quality of the Finnish measurements of precipitation and air quality at the four EMEP stations has been high during the whole measuring period. All these data are classified in the best A class (uncertainty within ±10%), except the NO2 data, which are classified in class C (uncertainty within ±30%). The detection limit of nitrogen dioxide is rather high compared to the low background concentrations at the stations, which leads to a higher uncertainty in the concentration.
2.2. Data checking
For the previous trend calculation of the FMI data, extending up to 1996, the time series in the selected primary data set were checked using both statistical methods to reveal anomalous values and individual inspection of these values.9 The objective of the data checking was to reject obvious errors e.g. contaminated values, but not to extract other high concentrations resulting from long range transport etc. For exposing suspect data, the time series were first divided into subsets according to season. Internal consistency of the subsets was tested by listing the values outside three times the standard deviation of a log normal distribution. Outlying values in weekly, fortnightly and monthly sampling data sets were inspected and evaluated individually by comparisons with the time series graphs and the respective values at neighbouring stations. For precipitation data, use was also made of ion balances, ion sum testing, and the comparison of the calculated and measured conductivity values. If an obvious reason, such as contamination of the sample, an error in the pre-treatment of the sample, or an error in the analysis, was found the outlier value was rejected; otherwise it was kept in the data set. All nitrogen dioxide 1 h concentration values that were more than four standard deviations away from the mean value or suspect according to the visual inspection were rejected.For this work, the results of the previous data checking up to 1996 were utilized. During 1997–2000, careful checking of the data along the monitoring chain was a continuous and defined procedure in the Quality Assurance Plan of the laboratory of the FMI. Thus, for these years, the reliability of the few exceptionally high values in the sulfur and nitrogen concentrations had already been inspected, mainly by means of methods described above, and no statistical treatment was considered necessary. Because of the different data quality checking procedure for the two time periods, there might be slight inequality in the data set. However, the effect of this possible inequality on the results is presumably very small, because the non-parametric trend calculation used in this work is not sensitive to occasional high values,16 and because mean values for 5 years were used when studying the accumulated exposure.
2.3. Trend calculations
The basic data for the trend analysis were the annual precipitation weighted concentrations in precipitation and the annual average concentrations of gas and aerosol components in air. The existence of a monotonic increasing or decreasing trend was tested with the non-parametric Mann-Kendall test16 at significance levels p < 0.05, p < 0.01, and p < 0.001 as a two-tailed test. Secondly the estimate for the slope of a linear trend was calculated with the non-parametric Sen's method.16,17 Sen's slope estimator is the median of the slopes calculated from all pairs of values in the data series. The 95% confidence interval for the slope estimator was also calculated.An estimate for the total change (ΔC) of annual values was calculated using Sen's slope estimator (Q) extracted from the annual values, and assuming a linear trend over the length of the period (Δt)
| ΔC = QΔt | (1) |
| Ci = Qti + B | (2) |
The relative total change indicated as a percentage is
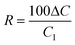 | (3) |
The total reduction used in the text and tables is −R.
In cases with a downward trend, when the slope is larger at the beginning, the method may overestimate the total relative change of annual values. The Sen's estimator was calculated only for time series with a significant trend of at least the p < 0.05 level.
2.4. Sectoral analysis
For the atmospheric concentrations at the EMEP stations, mean values and changes in time were studied also by an analysis of the transport sectors. The origin of each 24 h mean concentration of the gaseous and particulate sulfur and nitrogen compounds was calculated using the 2-dimensional 925 hPa trajectories obtained from the EMEP MSC-W.18 The trajectories calculations are based on meteorological data from the Numerical Weather Predicition models, LAM150 for 1985–1996 and PARLAM-PS for 1997–2000, both developed at the Norwegian Meteorological Institute. The 96 h backward trajectories were calculated by tracking the air parcel every 2 h for 96 h backwards in time, 4 times per day along modelled wind fields. The area around the arrival point was divided into 8 equal sectors. The criterion for the allocation of the trajectories of a particular arrival day to a specific sector was that at least 50% of their given positions during transport were found within that sector. If this criterion was not fulfilled, the sector for that day was set undetermined.The accumulated exposure hours of atmospheric sulfur and nitrogen from different sectors were calculated by multiplying the sector mean concentration by the number of days with transport from the corresponding sector. The sum was further multiplied by 24 h per day to give the unit of exposure, mg h m−3. In analysing the differences in exposures between the seasons, the months from October to March were included in the winter season, and the months from April to September in the summer season. Conventionally the term exposure is used from a medical or an environmental health point of view to describe the dose to human population. In this paper, however, the object of the exposure is natural environments, and the term is used in a similar sense as e.g. in the ozone exposure calculations for vegetation. Our exposure values are time-weighted airborne concentrations classified according to the air transport sector, which enables the comparison of different contributing directions.
2.5. Emissions and emission reductions in Europe
The atmospheric sulfur and nitrogen concentrations in Finland are not only a result of the national emissions but also affected by European wide emissions and their changes. The European emissions19 and emission reductions are summarized in Table 1. In this summary the emissions from the twenty-five European countries contributing most to the deposition of oxidized sulfur, oxidized nitrogen and reduced nitrogen in Finland in 2000 are included. The major contributors were taken from the calculations based on the Unified EMEP model of the EMEP/MSC-W.20 These selected countries are responsible for 94%, 99% and 98% of the country attributable oxidized sulfur, oxidized nitrogen and reduced nitrogen deposition, respectively, in Finland in 2000.| Emission area | Emission in 2000 kt | Emission reduction | |
|---|---|---|---|
| Period 1981–2000, % | Period 1990–2000, % | ||
| SO2 | |||
| Finland | 74 | 86 | 72 |
| Kola Peninsula | 280 | 73 | 57 |
| Russia ( Kola Peninsula excluded) | 1700 | 72 | 57 |
| Countries to the south of Finland | 5300 | 63 | 56 |
| Countries to the southwest and west of Finland | 5500 | 78 | 68 |
| Total | 13![[thin space (1/6-em)]](https://www.rsc.org/images/entities/i_char_2009.gif) 000 000 | 73 | 62 |
| NO2 | |||
| Finland | 236 | 15 | 21 |
| Kola Peninsula | 36 | −33 | 36 |
| Russia ( Kola Peninsula excluded) | 2300 | 39 | 35 |
| Countries to the south of Finland | 2500 | 44 | 43 |
| Countries to the southwest and west of Finland | 9600 | 28 | 27 |
| Total | 15000 | 33 | 32 |
| NH3 | |||
| Finland | 33 | 17 | 13 |
| Kola Peninsula | negligible | negligible | negligible |
| Russia | 650 | 45 | 45 |
| Countries to the south of Finland | 1300 | 45 | 42 |
| Countries to the southwest and west of Finland | 3200 | 11 | 9 |
| Total | 5200 | 28 | 35 |
The countries were further prearranged to four categories according to their geographic location with respect to Finland (see also Fig. 1). To the northeast of Finland, the only major emitters within the EMEP region are the metallurgic complexes on the Russian Kola Peninsula. The emissions of this region are provided apart from those of the rest of Russia, the latter situated to the east and southeast of Finland. The emissions from the Kola Peninsula were derived from the UNECE/EMEP gridded emission database.21
The continental East European countries located to the south of Finland compose the next category: Poland, Estonia, Ukraine, Romania, Hungary, Bulgaria, Belarus, Lithuania, Slovakia and Latvia.
The fourth category includes the countries to the southwest and west of Finland: Germany, United Kingdom, Sweden, Czech Republic, France, Spain, Italy, Belgium, Netherlands, Norway, Denmark, Ireland, Austria and Switzerland.
3. Results and discussion
3.1. Concentration trends
Because of the remarkable long range transport of sulfur to Finland,8 its concentrations are controlled by emissions from both Finland and the nearby countries. The Finnish SO2 emissions decreased in 1981–2000 even steeper than the concentration of any of the sulfur components, but the emissions of countries largely contributing to sulfur concentration and deposition in Finland declined neither as powerfully nor in phase with the Finnish emissions24(Table 1). Accordingly, the later turning point of the SO2 concentration at the southeastern and eastern stations compared to stations in the west is most probably a result of the slower emission reductions in eastern Europe.
Table 2 shows the trend statistics for the sulfur compounds for the periods 1981–2000 and 1990–2000. There were more stations in operation during the latter period. For the period 1981–2000, the trend was highly significant for all stations and compounds. The decline of the sulfur dioxide concentration since 1981 was dramatic. The concentration of this gaseous primary discharge component has decreased most, from 85% to over 95% in all areas of the country except northern Lapland, where a reduction of 65% has taken place. The impact of the large and slowly declining emissions from the Kola Peninsula is at least partly to be blamed for this lower reduction −90% of the sulfur deposition in the far north of Finland originated outside the country in 1987–1995.8 The slope estimate of sulfur dioxide is more uncertain than that of the other sulfur compounds because of its non-linear time series. However, it gives a rough indication of the relative total change.
| Area | Station | Period 1981–2000 | Period 1990–2000 | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Trend | Slope per year | Reduction | Trend | Slope per year | Reduction | ||||||
| Est. | Min. | Max. | per period, % | Est. | Min. | Max. | per period, % | ||||
| a n.s. not significant.b Period end 1999.c Years 1996–1998 missing.d Period 1992–1999. | |||||||||||
| SO2/µg S m−3 | |||||||||||
| SW | Utö | *** | −0.173 | −0.242 | −0.101 | >95 | * | −0.043 | −0.077 | −0.012 | 48 |
| SW | Guttorpb | n.s. | |||||||||
| SE | Virolahti | *** | −0.178 | −0.236 | −0.130 | 85 | ** | −0.130 | −0.192 | −0.067 | 63 |
| SE | Punkaharjub | *** | −0.168 | −0.251 | −0.103 | 85 | ** | −0.141 | −0.251 | −0.092 | 65 |
| C | Ähtäri | *** | −0.096 | −0.130 | −0.065 | >95 | ** | −0.051 | −0.093 | −0.020 | 70 |
| C | Sotkamob | *** | −0.171 | −0.210 | −0.120 | 94 | * | 81 | |||
| SL | Sodankyläb | *** | −0.100 | −0.138 | −0.067 | 84 | * | −0.067 | −0.164 | −0.002 | 67 |
| SL | Oulanka | ** | −0.049 | −0.084 | −0.024 | 55 | |||||
| NL | Kevob | ** | −0.069 | −0.123 | −0.037 | 64 | * | −0.078 | −0.178 | −0.005 | 53 |
| SO42− in aerosols/µg S m−3 | |||||||||||
| SW | Utö | *** | −0.051 | −0.061 | −0.036 | 61 | ** | −0.044 | −0.069 | −0.020 | 42 |
| SE | Virolahti | *** | −0.065 | −0.079 | −0.048 | 65 | ** | −0.071 | −0.100 | −0.036 | 51 |
| C | Ähtäri | *** | −0.040 | −0.049 | −0.029 | 63 | ** | −0.034 | −0.053 | −0.013 | 44 |
| SL | Oulanka | ** | −0.024 | −0.036 | −0.008 | 39 | |||||
| SO42− in precipitation/mg S l−1 | |||||||||||
| SW | Utö | *** | −0.063 | −0.079 | −0.048 | 69 | ** | −0.057 | −0.073 | −0.020 | 51 |
| SW | Guttorpb | ** | −0.034 | −0.061 | −0.018 | 40 | |||||
| SW | Kotinen | ** | −0.039 | −0.055 | −0.018 | 54 | |||||
| SE | Virolahti | *** | −0.051 | −0.069 | −0.038 | 62 | * | −0.051 | −0.082 | −0.024 | 49 |
| SE | Punkaharju | *** | −0.035 | −0.044 | −0.022 | 67 | * | −0.028 | −0.045 | −0.014 | 42 |
| C | Ähtäri | *** | −0.027 | −0.033 | −0.021 | 68 | ** | −0.023 | −0.038 | −0.018 | 48 |
| C | Hietajärvi | ** | −0.033 | −0.045 | −0.020 | 53 | |||||
| C | Sotkamo | *** | −0.031 | −0.042 | −0.020 | 69 | * | −0.011 | −0.032 | −0.002 | 25 |
| C | Hailuoto | *** | −0.033 | −0.043 | −0.022 | 66 | ** | −0.041 | −0.064 | −0.024 | 61 |
| SL | Oulanka | ** | −0.022 | −0.031 | −0.010 | 57 | |||||
| SL | Pesosjärvib | ** | −0.036 | −0.044 | −0.020 | 62 | |||||
| SL | Sodankylä | *** | −0.026 | −0.034 | −0.015 | 75 | ** | −0.017 | −0.033 | −0.007 | 46 |
| NL | Värriöc | * | −0.017 | 42 | |||||||
| NL | Kevo | *** | −0.020 | −0.022 | −0.015 | 69 | n.s. | ||||
| NL | Vuoskojärvib | n.s. | |||||||||
| NL | Särkijärvid | n.s. | |||||||||
In the 1990's, the overall downward trend continued, but the significances were lower. The shorter time period, with fewer annual values in the test, partly reduced the significance of the result.
Guttorp in the southwest is the only station where the SO2 concentration has hardly declined during the 1990's and shows no significant trend. The average concentration for the summer months decreased slowly until 1996 and the average winter concentration fluctuated around 1.0 µg m−3. During 1997–1998, the summer concentrations showed a tendency to increase, approaching the level of the winter concentrations. The dominant wind directions during the summer months in Guttorp are south and southwest (sum 36%, mean value in 1971–2000) as well as north (21%).25 During 1997–1998, however, the proportion of the northern winds was about 25% lower and the proportion of the eastern and southeastern winds about 45% higher than the long time average. An increased transport from the east and southeast was also obtained in 1997–1998 in the trajectories arriving in Utö (SW). Because the highest mean concentrations of SO2 are observed in air arriving from the east and southeast, the increased summer concentrations in 1997–1998 in Guttorp are explained by the larger proportion of more polluted air from these transport directions. For the total monitoring period for Guttorp (1985–1999), the downward trend of SO2 was highly significant, with a total reduction of 79%.
The concentrations of the other sulfur compounds have declined rather evenly since 1981. The downward trend of sulfate in particles was statistically significant in all areas where monitoring was conducted. The overall reduction was about 60% during 1981–2000, and 40–50% during 1990–2000.
Finally, the downward trend in the concentration of sulfate in precipitation was highly significant in 1981–2000, and a total decline of 60–75% was calculated for all stations with the long time series. In the 1990's, the concentration continued to decline even if the significance of the trend was lower. The total reduction per period was 40–60%, except for Sotkamo (C) where only a 25% reduction was observed. At the northernmost stations there was no trend except for the northeastern Värriö (NL). However, the mean concentration at all stations in northern Lapland has decreased from around 0.3 mg l−1 to around 0.2 mg l−1 during the 1990's. The sulfate deposition in northern Finland is of highly episodic character—a few long-range transport episodes often dominate the annual deposition.26 Thus, coincidental meteorological factors impact strongly the annual mean concentration of oxidized sulfate in this area of Finland, and a decade might not be long enough to detect a trend.
Sulfur dioxide emissions have declined strongly in all emission areas contributing to the long-range transport to Finland (see Table 1). The domestic emissions in Finland have declined most, contributing positively on the sulfur dioxide trend. However, the trends of the sulfate compounds are lower than those of sulfur dioxide, which is partly explained by the slower cut down of the large emissions in the important southern sector. The sulfate concentrations in air and precipitation all over the country have, roughly speaking, declined approximately as much as the mean of the sulfur dioxide emissions in Table 1.
Table 3 shows the trend statistics for all the nitrogen compounds for the period 1990–2000. For the components in precipitation the trends could also be calculated for the period 1981–2000.
| Period 1981–2000 | Period 1990–2000 | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Trend | Slope per year | Reduction | Trend | Slope per year | Reduction | ||||||
| Area | Station | Est. | Min. | Max. | per period, % | Est. | Min. | Max. | per period, % | ||
| a n.s. not significant.b Year 1996 missing.c Period 1990–1999.d Years 1996–1998 missing.e Period 1992–1999. | |||||||||||
| NO3− gas + aerosol/µg N m−3 | |||||||||||
| SW | Utö | n.s. | |||||||||
| SE | Virolahti | * | −0.009 | −0.018 | −0.002 | 25 | |||||
| C | Ähtäri | n.s. | |||||||||
| SL | Oulanka | ** | −0.003 | −0.004 | −0.002 | 33 | |||||
| NH4+ gas + aerosol/µg N m−3 | |||||||||||
| SW | Utö | n.s. | |||||||||
| SE | Virolahti | n.s. | |||||||||
| C | Ähtäri | *** | −0.030 | −0.038 | −0.018 | 50 | |||||
| SL | Oulanka | n.s. | |||||||||
| NO2/µg N m−3 | |||||||||||
| SW | Utöb | n.s. | |||||||||
| SE | Virolahtib | n.s. | |||||||||
| C | Ähtäri | n.s. | |||||||||
| SL | Oulanka | n.s. | |||||||||
| NO3− in precipitation/mg N l−1 | |||||||||||
| SW | Utö | ** | −0.017 | −0.028 | −0.007 | 39 | n.s. | ||||
| SW | Guttorp | n.s. | |||||||||
| SW | Kotinen | n.s. | |||||||||
| SE | Virolahti | * | −0.005 | −0.013 | 0.000 | 20 | n.s. | ||||
| SE | Punkaharju | *** | −0.007 | −0.011 | −0.004 | 36 | n.s. | ||||
| C | Ähtäri | *** | −0.007 | −0.009 | −0.004 | 37 | * | −0.006 | −0.012 | −0.002 | 23 |
| C | Hietajärvi | n.s. | |||||||||
| C | Sotkamo | n.s. | n.s. | ||||||||
| C | Hailuoto | n.s. | |||||||||
| SL | Oulanka | n.s. | |||||||||
| SL | Pesosjärvic | n.s. | |||||||||
| SL | Sodankylä | * | −0.004 | −0.007 | −0.001 | 35 | n.s. | ||||
| NL | Värriöd | ** | −0.006 | 39 | |||||||
| NL | Kevo | * | −0.002 | −0.004 | −0.001 | 32 | n.s. | ||||
| NL | Vuoskojärvic | n.s. | |||||||||
| NL | Särkijärvie | n.s. | |||||||||
| NH4+ in precipitation/mg N l−1 | |||||||||||
| SW | Utö | *** | −0.026 | −0.034 | −0.015 | 61 | ** | −0.021 | −0.043 | −0.005 | 39 |
| SW | Guttorp | n.s. | |||||||||
| SW | Kotinen | * | −0.010 | −0.027 | −0.001 | 36 | |||||
| SE | Virolahti | *** | −0.022 | −0.032 | −0.014 | 56 | n.s. | ||||
| SE | Punkaharju | ** | −0.013 | −0.021 | −0.005 | 63 | n.s. | ||||
| C | Ähtäri | *** | −0.011 | −0.015 | −0.006 | 60 | * | −0.009 | −0.021 | −0.003 | 39 |
| C | Hietajärvi | * | −0.012 | −0.020 | −0.002 | 53 | |||||
| C | Sotkamo | ** | −0.008 | −0.014 | −0.004 | 59 | n.s. | ||||
| C | Hailuoto | n.s. | |||||||||
| SL | Oulanka | n.s. | |||||||||
| SL | Pesosjärvic | * | −0.007 | −0.012 | 0.000 | 50 | |||||
| SL | Sodankylä | *** | −0.009 | −0.013 | −0.005 | 80 | n.s. | ||||
| NL | Värriöd | n.s. | |||||||||
| NL | Kevo | ** | −0.005 | −0.009 | −0.002 | 84 | n.s. | ||||
| NL | Vuoskojärvic | n.s. | |||||||||
| NL | Särkijärvie | n.s. | |||||||||
For the gas phase and particulate nitrogen compounds, the measurements covered the 1990's. The statistical test gave few significant trends: the sum of nitrate and nitric acid declined by around 30% at stations in the southeast and in southern Lapland, and the sum of ammonia and ammonium decreased by 50% in the southern part of central Finland. The significance of the ammonium trend was, however, very high (p < 0.001). No significant trend was detected for nitrogen dioxide. The test accepts the missing values in the time series of nitrogen dioxide, but they weaken the test and might prevent exposing trends with weak significance. Further, the change in the monitoring method of nitrogen dioxide in 1996 increased the uncertainty in the calculation of the trend over that period. On the other hand, the result of no trend illustrates well the modest reduction in the domestic nitrogen dioxide emissions, which highly affect the concentration.
The components NH3/NH4+, SO2/SO42− and NO2/NO3− interact with each other in the secondary aerosol formation resulting in differences in the formation of fine and coarse particulate matter and — due to their different sizes — different atmospheric lifetimes and consequently different long range transport. Furthermore different precursor compositions will lead to different rates of aerosol formation. Sulfate and ammonium react readily and are mainly found in fine particles, they might be transported to Finland from a long distance, but also from nearby areas.4 Large part of the nitrate is carried to Finland with maritime air masses in coarse particles, which deposit faster and cannot be of distant origin.27 The formation of NH4NO3 (in fine particles) needs excess ammonia compared to sulfate4 and is favoured by cool and humid conditions.27 Thus, many factors, besides the primary emissions, affect the observed concentrations and trends of these compounds, including their relative concentrations in the atmosphere, the reversible nature of the reactions and the meteorological situation.
According to the statistical test, the downward trend of the concentration of nitrate in precipitation during 1981–2000 was highly significant at most stations in the south and at Ähtäri (C) in the southern part of central Finland. At these stations the total decline per period reached 40%. At Virolahti (SE), in the southeastern corner of Finland, the concentration has undergone a rather low total reduction of 20%. The reason might be different transport routes from areas with slower emissions reductions. Finally, the significance of the trend was lower in the areas in the north, but the calculated total reduction was almost as high as in the south. For 1990–2000 the calculations resulted in only very few significant trends, indicating minor domestic and European emission reductions and fluctuation of the concentrations.
The ammonium concentration in precipitation has declined significantly between 1981 and 2000. The statistical test gave as high probabilities for the existence of trends as for the sulfur compounds. Also the total reductions follow very regularly those of sulfate with only slightly lower values. The overall reduction of ammonium in precipitation during 1981–2000 was around 60% in the southern and central parts of the country, and 80% in the north.
The higher reduction of ammonium in precipitation in the north might contain a larger uncertainty than the values obtained for nitrate in precipitation. Firstly, ammonium is usually transported for shorter distances than nitrate,28 and possible local and regional sources, like agriculture, have a relatively larger effect on the concentration. Secondly, the concentration, especially in the north, was so low that even minor ammonium sources not known to us could have weakened the reliability of the time series. Finally, the ammonium concentration in the samples is contaminated more easily than the other compounds. This may have occurred in the beginning of the monitoring period in spite of the careful data checking, which would lead to too high annual mean values.
In the 1990's, there was a downward trend of ammonium in precipitation at two of the three stations in the southwest and in the southern part of central Finland (reduction ca. 40%). In the southeast the decline did not continue. In the east (Hietajärvi) and at one of the southern Lapland stations (Pesosjärvi), a reduction of 50% was reached.
The domestic nitrogen dioxide emissions have declined only by 15–20%, which might be too low to be by itself detected by the monitoring. From the Finnish perspective, the nitrogen dioxide emissions are highest in the countries in the southwestern and western sectors. There the reduction has been lower than the average in the area contributing to Finland (Table 1). The frequent transport from these areas has highly affected the Finnish concentrations. Nitrate in the precipitation has declined approximately as much as the mean of the nitrogen dioxide emissions in Table 1, and more than the domestic emissions.
The Finnish annual ammonia emissions have been reduced by 17% during 1981–2000 (Table 1). So the reductions of the ammonium concentration in precipitation are clearly larger than the reduction of the national ammonia emissions. The ammonium emissions in other countries have declined most, by 45%, in the east and southeast sectors and in the south (Table 1). The largest emissions, from the Finnish perspective, are in countries in the southwestern and western sectors, where also the reduction has been lower than elsewhere. This indicates that the relative importance of these sectors to the ammonium load might have increased with time. The ammonium concentration in precipitation in Finland has declined much more than the emissions on average or in any of the areas in Table 1. This could be explained by a larger reduction of the long range transported ammonium sulfate caused by the international sulfur reductions.
Similar results have been reported from monitoring covering many other western and central European countries.30–32 Decreasing sulfur trends have been measured also at high elevations, e.g. at most of the studied 11 Austrian background stations, covering altitudes from 150 to 3100 m, a statistically significant decrease in SO42− in precipitation was observed in 1990–1997, whereas again for the nitrogen compounds fewer trends were provided.33 In North America, several studies show, that the sulfur concentrations in air and precipitation were highest in the 1980's and have decreased since then; in contrast, the nitrate concentrations generally have not changed as much.34–37
3.2. Exposures and their trends in relation to air transport sector
Daily concentrations of gas phase and particulate sulfur and nitrogen components together with the estimates of air transport routes enable a sectoral examination of the pollution exposure. The sectoral exposure means here the sum of the daily loads arriving from a specific sector over the time period assessed. It is counted as mg h m−3. Further, the accumulated annual exposure would be the sum of the summer and winter exposures in all eight sectors plus the approximately 30% of the daily exposure values that could not be classified to a specific transport sector.The sectoral distributions in Figs. 2 and 3 illustrate roughly the relative importance of different transport directions to the sulfur and nitrogen dry deposition in Finland even if absolute dry deposition values cannot be calculated. It can be assumed that nearby sources and the background concentration found everywhere maintain a low basic value for all compounds in all of the sectors, especially during low wind velocities. However, the monitoring stations have been located far from any larger local sources. Thus, it can be supposed that the differences between the sectors originate mainly from loads transported from outside the immediate surroundings of the station, which can be separated rather well with the classification method used in this paper. Since the concentration data is 24 h mean values, it cannot be combined with wind velocity observations which might change largely within 24 h.
 | ||
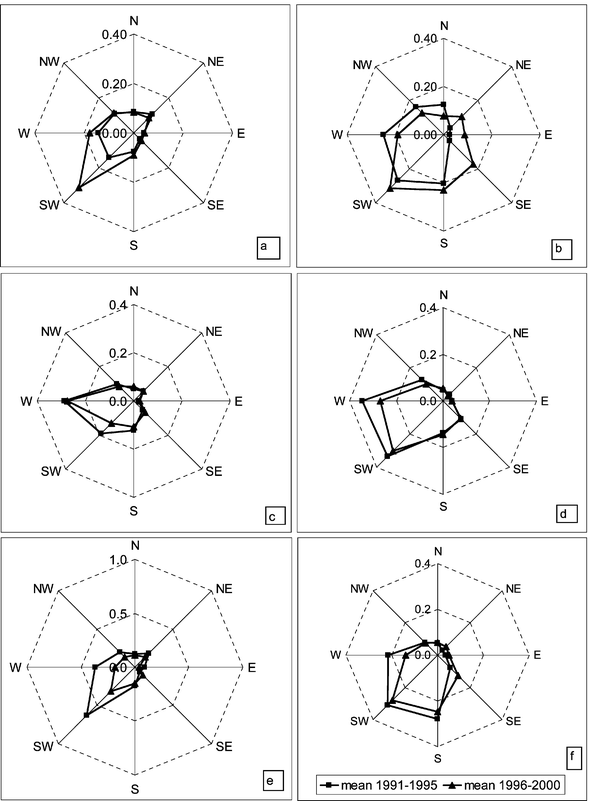
| Fig. 2 Five year mean values for total atmospheric sulfur exposure from different sectors, sum of summer or winter season. Unit mg h m−3. Stations, regions and seasons: (a) Utö, SW, summer; (b) Utö, SW, winter; (c) Virolahti, SE, summer; (d) Virolahti, SE, winter; (e) Ähtäri, C, summer; (f) Ähtäri, C, winter; (g) Oulanka, SL, summer; (h) Oulanka, SL, winter. | ||
 | ||
| Fig. 3 Five year mean values for nitrogen exposure from different sectors, sum for summer or winter season. Unit mg h m−3. Component, station, area and season: (a) NO2–N, Oulanka, SL, summer; (b) NO2–N, Oulanka, SL, winter; (c) (HNO3 + NO3−)–N, Virolahti, SE, summer; (d) (HNO3 + NO3−)–N, Virolahti, SE, winter; (e) (NH3 + NH4+)–N, Ähtäri, C, summer; (f) (NH3 + NH4+)–N, Ähtäri, C, winter. | ||
The distribution patterns, as 5-year mean values for 1986–2000, are given for the four stations measuring daily values. The accumulated summer and winter 5-year mean values are discussed separately because of seasonal differences in the frequency of the transport sectors. In summer, the gaseous and particulate sulfur influenced the total sulfur exposure with rather equal weights. In the winter exposure, the weight of gaseous sulfur was about two thirds.
In winter, all sectors towards the south were important to the sulfur exposure. At Oulanka (SL), the importance of transport from the northeast and southeast has lately increased (Fig. 2h), presumably because of the rather low emission reduction in the NE sector (Table 1). At Virolahti (SE), the mean concentrations of SO2 in air masses arriving from the southeast and south sectors were elevated compared to air in the other sectors during the whole period investigated (Fig. 2d). Although transport from these sectors was not very frequent, its weight to the wintertime exposure – and dry deposition – has been and is still evident. Differences between the seasons are in line with large-scale meteorological observations in southeastern Finland: in summer the southwesterly flow dominates and the effect of the elevated concentrations of sulfur dioxide in the southeastern air masses evens out, while in winter there is no remarkable difference between the southwesterly, southerly and southeasterly winds.25
In general, the annual exposure of sulfur has declined strongly in the 1980's; reduction has taken place in almost all sectors. In the 1990's this positive development has continued in the transport from the sectors towards west and south (S, SW, W, and NW). Emissions have decreased most in countries to the SW and W of Finland, whereas the likewise high emissions in the S sector have declined less (Table 1). In contrast, the exposure from most of the sectors towards east and north (N, NE, E, and SE) has either stayed unchanged or increased during the last decade. For instance, at Oulanka (SL), the transport from the sectors ranging from the north to the southeast has risen 20%, whereas the transport from the sectors ranging from the south to the northwest has decreased by 40% in the 1990's (Fig. 2g, h). Thus, the effect of the transport from east and north has slightly increased.
The wintertime exposures have decreased more than the summertime exposures. This is in line with the observation that the downward trend in the winter mean concentration of sulfur dioxide was higher than the trend in the summer mean concentration.9 In the exposure, the largest decline has taken place already between the first two 5-year periods (1986–1990 and 1991–1995). After that, many sectors with low exposure values have remained unchanged although the emissions in all the sectors have continued to decline in the 1990's. This can be seen especially at Utö (SW), where only the frequent western and southwestern transport with a rather low mean concentration has continued to decline after 1991–1995 (Fig. 2a, b). Thus, the present decrease of sulfur exposure at Utö is mainly controlled by concentration changes in the transport from these sectors; the frequency of the transport sectors has not changed much during the assessed period.
Our sectoral exposure estimates at Oulanka (SL) confirm the earlier estimates by Tuovinen et al.38 that in summer the total accumulated dry deposition there was dominated by transport from north and northeast, while in winter the influence of the southern sectors increased. At Sevettijärvi in northern Lapland in the vicinity of the Kola Peninsula emission sources, transport from east and northeast was dominant for SO2 and transport from southwest for SO42−, when the exposure was averaged over a two year period.39 The sectoral exposure estimates given in this paper are in line with calculations by the EMEP model of export from other countries to Finland; the largest foreign contribution to the sulfur deposition comes from Russia, Germany, Poland and the United Kingdom.24
The estimate of the NO2 exposure is more uncertain than the other estimates, because of more missing values and higher uncertainties in the NO2 data series. Because of too many missing daily values, no 5-year mean exposure for 1991–1995 could be calculated from the Virolahti (SE) and Utö (SW) data. At Oulanka (SL), the exposure from the eastern transport sectors in winter increased notably between the two 5-year periods (1991–1995 and 1996–2000) whereas in summer the transport peaked in the southwest sector (Fig. 3a,b). On the annual NO2 exposure at Oulanka, even the northern transport sectors had a remarkable effect. Thus, a slight shift towards increased transport from the eastern and northern sectors appears possible. Increased emissions from traffic were most probably the reason for the higher exposures.
Total atmospheric nitrate (Fig. 2c, d) accumulated strongly with transport by southwesterly and westerly air masses arriving from sectors, where the NO2 emissions were the largest. In winter the south and southeast sectors were also important. The emission reduction was largest in the countries in the S sector. The sectoral exposure from N, NE, E, SE and S did not change much between the periods at Virolahti (SE), the largest reductions were in transport from southwest in summer and west in winter. At Oulanka (SL) the exposure decreased in the west and northwest sectors in winter.
Finally, the pattern of exposure of the total atmospheric ammonium (Fig. 2e, f) greatly resembled the corresponding distribution for particulate sulfate; the transport from southwest peaked during summer and the transport from west, southwest and south dominated during winter. This is explained by the well known transport of particles containing ammonium sulfate. The winter exposure did not change much, in summer the contribution from west and southwest declined sharply at Ähtäri (C). It is supposed, that the domestic NH3 emissions and their low reduction have highly affected the NH3 exposure. However, also important have been the largest emissions in the SW and W sectors, and their low decline.
4. Summary and conclusions
In Finland, the SO2 emission and concentrations in background areas have decreased sharply since 1981, 86% and ca. 85–95%, respectively. For sulfate concentrations in air and precipitation reductions have been lower due to the dominance of the long range transport. From the Finnish perspective, the emissions of SO2 are largest in the countries to the south, and southwest and west of Finland. The frequent transport from these sectors together with the lower SO2 emission reduction, especially in the southern sector, has contributed to the lower reductions in the sulfate concentrations. In southeastern Finland the decline of the SO2 concentration started later than in other parts of the country, because of the larger contribution of southeastern transport from areas with slower emission reductions. In the northernmost part of the country no significant downward trend could be detected in the 1990's for sulfate in precipitation, and the reduction in the SO2 concentration since 1981 was lower than elsewhere in the country, which deserves attention when estimating the changes in the critical load of acidity.The Finnish and European NOx emissions have decreased during 1991–2000 21% and 32%, respectively. These reductions are reflected in the non-decreasing or only slightly decreasing concentrations of the oxidized nitrogen compounds. However, the concentration of ammonium in precipitation has decreased significantly during 1981–2000, more than the Finnish and European NH3 emissions. Monitoring of the background concentrations of the nitrogen compounds should thus be continued all over the country in order to verify possible changes in their time series.
The sulfur exposure in Finland was highly affected by transport from the southeastern, southern and southwestern sectors. At Oulanka in southern Lapland the contribution from the northern and northeastern sectors was also remarkable. During the summer months, transport from the southwestern and western sectors dominated, while in winter the distribution was more even, with higher values from all the sectors towards the south. Remarkable for the sulfur exposure is the change in the weight of the transport sectors in the 1990's: exposure from the west and south has still declined, whereas transport from the north and east has turned to a slight increase.
The nitrogen exposure was also mainly dominated by transport from sectors between west and south. The emissions of NOx and NH3 are highest in the countries to the southwest and west of Finland. The rather small changes in the exposure patterns during 1991–2000 reflected the minor reductions of the NOx emissions. Also for the nitrogen exposure, there seems to have been a slight transition towards increased eastern and northern contribution, even if the total exposure was mostly affected by transport from the southern and western sectors.
References
- Acidification in Finland, ed. P. Kauppi, P. Anttila and K. Kenttämies, Springer-Verlag, Berlin, 1990 Search PubMed.
- K. Barrett, J. Schaug, A. Bartonova, A. Semb, A.-G. Hjellbrekke and J. E. Hanssen, A Contribution from CCC to the Reevaluation of the observed Trends in Sulfur and Nitrogen in Europe 1978–1998, EMEP/CCC-Report 7/2000, Norwegian Institute for Air Research, Norway, 2000 Search PubMed.
- J. H. Seinfeld and S. N. Pandis, Atmospheric Chemistry and Physics: From Air Pollution to Climate Change, John Wiley & Sons, Inc., New York, 1997 Search PubMed.
- T. A. Pakkanen, R. E. Hillamo, M. Aurela, H. V. Andersen, L. Grundahl, M. Ferm, K. Persson, V. Karlsson, A. Reissell, O. Røyset, I. Fløisand, P. Oyola and T. Ganko, J. Aerosol. Sci., 1999, 30, 247 CrossRef CAS.
- T. A. Pakkanen, R. E. Hillamo, M. Mäkinen, T. Mäkelä, V.-M. Kerminen, A. Virkkula, V. Karlsson, A. Reissell, H. Lättilä, H. Boij, G. Hilbert, L. Grundahl, H. V. Andersen, L. Christensen, M. Ferm, K. Persson, I. Fløisand, O. Røyset, J. Schaug, K. Tørseth, P. Oyola, T. Ganko, H. Carlsson, K. Rosman, V. Vesely and L. Granat, Nordic HNO3/NO3− and NH3/NH4+ gas/particle intercomparison in Helsinki, Publications on Air Quality No. 18, Finnish Meteorologial Institute, Helsinki, 1994 Search PubMed.
- M. Forsius, S. Kleemola, J. Vuorenmaa and S. Syri, Water, Air, Soil, Pollut., 2001, 130, 1641 CrossRef.
- L. Ukonmaanaho, M. Starr and T. Ruoho-Airola, Water, Air, Soil, Pollut., 1998, 105, 353 CrossRef CAS.
- T. Ruoho-Airola, S. Syri and G. Nordlund, Boreal Env. Res., 1998, 3, 205 Search PubMed.
- A. Kulmala, L. Leinonen, T. Ruoho-Airola, T. Salmi and J. Waldén, Air Quality Trends in Finland, Air Quality Measurements, Finnish Meteorological Institute, Helsinki, 1998 Search PubMed.
- K. Tørseth, A. Wenche and S. Solberg, Water, Air, Soil, Pollut., 2001, 130, 1493 CrossRef.
- K. Kindbom, A. Svensson, K. Sjöberg and G. Pihl Karsson, Trends in Air Concentration and Deposition at Background Monitoring Sites in Sweden, IVL Report B 1429, 2001 Search PubMed.
- EMEP/CCC, EMEP Manual for Sampling and Chemical Analysis. http://www.nilu.no/projects/ccc/manual/index.html.
- Environment Data Centre, Manual for Integrated Monitoring. Environmental Report 5, National Board of Waters and the Environment, Helsinki, 1993 Search PubMed.
- Air Quality Measurements 2000, ed. L. Leinonen, Finnish Meteorological Institute, Helsinki, 2001 Search PubMed.
- http://www.emep.int/areas/index.html, 31.7.2003.
- R. O. Gilbert, Statistical Methods for Environmental Pollution Monitoring, Van Nostrand Reinhold, New York, 1987 Search PubMed.
- P. K. Sen, J. Am. Stat. Assoc., 1968, 63, 1379.
- http://www.emep.int/index_assessment.html, 31.7.2003.
- V. Vestreng, Review and Revision, Emission data reported to CLRTAP, EMEP/MSC-W Note 1/2003, Norwegian Meteorological Institute, Oslo, 2003 Search PubMed.
- L. Tarrasón, J. E. Jonson, H. Fagerli, A. Benedictow, P. Wind, D. Simpson and H. Klein, Transboundary acidification, eutrophication and ground level ozone in Europe, Part III, Source–Receptor relationship, EMEP Report 1/2003. Norwegian Meteorological Institute, Oslo, 2003 Search PubMed.
- WebDab, UNECE/EMEP emission database, http://webdab.emep.int/. 15.10.2003.
- A. Voipio and M. Leinonen, Itämeri (The Baltic Sea), Kirjayhtymä, Helsinki, 1984, p. 53 Search PubMed.
- CRC Handbook of Chemistry and Physics, ed. R. C. Weast, CRC Press, Cleveland, Ohio, 52nd edn., 1971, sect. F-165 Search PubMed.
- L. Tarrasón and J. Schaug, Transboundary Acidification and Eutrophication in Europe, EMEP Report 1/2000, Norwegian Meteorological Institute, Oslo, 2000 Search PubMed.
- A. Drebs, A. Nordlund, P. Karlsson, J. Helminen and P. Rissanen, Climatological Statistics of Finland 1971–2000, Climatic Statistics of Finland 2002:1, Edita Prima, Helsinki, 2002 Search PubMed.
- T. Ruoho-Airola and T. Salmi, Water, Air, Soil, Pollut., 2001, 130, 529 CrossRef.
- V.-M. Kerminen, T. A. Pakkanen and R. E. Hillamo, Atmos. Environ., 1997, 31, 2753 CrossRef CAS.
- L. Tarrasón, K. Barrett, L. Erdman and A. Gusev, Atmospheric Supply of Nitrogen, Lead and Cadmium to the Baltic Sea, EMEP/MSC-W Research Report no. 58, Norwegian Meteorological Institute, Oslo, 1997 Search PubMed.
- F. Moldan, R. F. Wright, S. Löfgren, M. Forsius, T. Ruoho-Airola and B. L. Skjelkvåle, Hydrol. Earth Syst Sci., 2001, 5, 339 Search PubMed.
- J. G. Irwin, G. Gampbell and K. Vincent, Atmos. Environ., 2002, 36, 2867 CrossRef CAS.
- F. Zimmermann, H. Lux, W. Maenhaut, J. Matschullat, K. Pressow, F. Reuter and O. Wienhaus, Atmos. Environ., 2003, 37, 171 CrossRef.
- G. Arends, J. H. Baard and H. M. Ten Brink, Atmos. Environ., 1997, 31, 4063 CrossRef.
- S. Tsakovski, J. Puxbaum, V. Simenov, M. Kalina, H. Löffler, G. Heimburger, P. Biebl, A. Weber and A. Damm, J. Environ. Monit., 2000, 2, 424 RSC.
- J. A. Lynch, V. C. Bowersox and J. W. Grimm, Environ. Sci. Technol., 2000, 34, 940 CrossRef CAS.
- B. B. Hicks, R. S. Artz, T. P. Meyers and R. P. Hosker, Jr., J. Geophys. Res., 2002, 107(10) DOI:10.1029/2000JD000165.
- D. M. Holland, P. P. Principe and J. E. Sickes, Atmos. Environ., 1999, 33, 37 CrossRef CAS.
- J. D. Shannon, Atmos. Environ., 1999, 33, 807 CrossRef CAS.
- J.-P. Tuovinen, T. Laurila, H. Lättilä, A. Ryaboshapko, P. Brukhanov and S. Korolev, Atmos. Environ., 1993, 27A, 1379.
- A. Virkkula, M. Mäkinen, R. Hillamo and A. Stohl, Water, Air, Soil, Pollut., 1995, 85, 1997 CAS.
| This journal is © The Royal Society of Chemistry 2004 |