Mono- and binuclear cyclometallated palladium(II) complexes containing bridging (N,O-) and terminal (N-) imidate ligands: Air stable, thermally robust and recyclable catalysts for cross-coupling processes
Ian J. S.
Fairlamb
*a,
Anant R.
Kapdi
a,
Adam F.
Lee
a,
Gregorio
Sánchez
b,
Gregorio
López
b,
José Luis
Serrano
c,
Luis
García
c,
José
Pérez
c and
Eduardo
Pérez
c
aDepartment of Chemistry, University of York, Heslington, York, UK YO10 5DD. E-mail: ijsf1@york.ac.uk; Fax: 44 (0)1904 432516; Tel: 44 (0)1904 434091
bDepartamento de Química Inorgánica, Universidad de Murcia, 30071, Murcia, Spain
cDepartamento de Ingeniería Minera, Geológica y Cartográfica. Área de Química Inorgánica, 30203, Cartagena, Spain
First published on 27th October 2004
Abstract
Novel dinuclear cyclometallated palladium complexes [{Pd(µ-NCO)(C⁁N)}2], containing asymmetric imidato –NCO– bridging units have been synthesised [C⁁N = 7,8-benzoquinolyl; –NCO– = succinimidate (1c), phthalimidate (2c) or maleimidate (3c)]. The reaction of these complexes, and the previously reported analogous imidate precursors containing a phenylazophenyl (1a–3a) or 2-pyridylphenyl (1b–3b) backbone, with tertiary phosphines provides novel mononuclear N-bonded imidate derivatives of the general formula [Pd(C⁁N)(imidate)(L)] [L = PPh3, P(4-F-C6H4)3 or P(4-MeO-C6H4)3]. The single crystal structures of [Pd(azb)(phthalimidate)(P(4-MeO-C6H4)3)] (9a) and [Pd(bzq)(phthalimidate)(PPh3)] (7c) have been established. Dinuclear complexes (1a–3a, 1b–3b, 1c–3c) demonstrate outstanding thermal stability in the solid-state, as shown by thermoanalytical techniques. A marked influence of bridging imidate groups on the initial decomposition temperature is observed. The dinuclear and mononuclear derivatives are shown to be active catalysts/precatalysts for the Suzuki–Miyaura cross-coupling reactions of aryl bromides with aryl boronic acids, and the Sonogashira reactions of aryl halides with phenyl acetylene (in the presence and absence of Cu(I) salts). The conversions appear to be dependent, to some extent, on the type of imidate ligand, suggesting a role for these pseudohalides in the catalytic cycle in both cross-coupling processes. Lower catalyst loadings in ‘copper-free’ Sonogashira cross-couplings favour higher turnover frequencies. We have further determined that these catalysts may be recycled using a poly(ethylene oxide) (PEO)/methanol solvent medium in Suzuki–Miyaura cross-coupling. Once the reaction is complete, product extraction into a hexane/diethyl ether mixture (1 ∶ 1, v/v) gives cross-coupled products in good yields (with purity > 95%). The polar phase can then be re-used several times without appreciable loss of catalytic activity.
Introduction
Over the last twenty-five years great advances in the development of palladium-catalysed carbon–carbon and carbon–heteroatom bond forming processes have been made.1 Key reactions, such as Sonogashira, Stille and Suzuki–Miyaura coupling,2 have made significant contributions to synthetic chemistry, not least in the synthesis of important natural products and materials.3 For this reason, the design of electron rich palladium complexes, possessing strong σ-donor ligands, that mediate mild cross-coupling reactions of activated and deactivated organohalide substrates with appropriate nucleophiles, has attracted continued attention.4 The use of cyclometallated Pd(II) complexes as precatalysts, has in part, led the advances seen in the last decade. Phosphapalladacycles and N-heterocyclic carbene Pd-complexes were first successfully introduced by Herrmann and co-workers, as general catalysts for C–C cross-coupling reactions, which demonstrate broad substrate scope.4a,4c Since then, less complicated systems, as well as “pincer” palladacycles, containing functional groups bound to Pd by N-, P- or S-atoms, have been shown to be efficient catalysts for Suzuki–Miyaura cross-coupling, amongst other coupling reactions.4d,5 With regard to N-donor palladacycles, Bedford6 and others7 have developed the use of dinuclear chloride bridged orthopalladated N,N-dimethylbenzylamine complexes in cross-coupling processes utilising aryl bromides. Related dinuclear orthometallated complexes containing imine8 and oxime groups also exhibit interesting catalytic activity; in the latter case, with deactivated aryl chlorides, where water and tetrabutylammonium bromide played a key role.9 Dinuclear C,N-palladacycles and the phosphine adducts, of the type [Pd(C⁁N)(PR3)(X)] [C⁁N = imine or amine based orthometallated ligand; X = Cl or trifluoroacetate (TFA)], exhibit increased catalytic activity in Suzuki–Miyaura cross-coupling.10 Notably, Bedford and co-workers have emphasised the enhanced activity of these palladacycles in comparison with conventional precursors. Crucially, they observed the important role played by both the phosphine and the anionic ligand “X” in the overall performance of catalysts (catalyst activity and lifetime). Understanding the role played by the anionic ligand has been of interest to us, particularly with respect to imidato ligands, such as maleimide and derivatives, phthalimide and succinimide. Imidato ligands offer, potentially, monodentate (N-coordination, as in I) or bidentate modes of coordination (N,O or N,N) (Fig. 1, as in II–IV). The hemilability associated with O-coordination might offer stabilisation of key catalytic intermediates.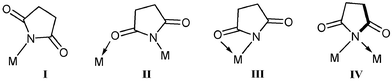 | ||
| Fig. 1 Possible coordination modes of succinimide ligands to metal centres. | ||
Indeed, as a part of ongoing studies into Pd- and Pt-imidato complexes, several synthetic routes to various complexes have been studied. The catalytic application of some of these new complexes has been described by our group in Stille and Suzuki–Miyaura cross-coupling reactions.11,12
The oxidative addition of N-bromosuccinimide (NBS) to Pd(0) and Pt(0) precursors in the presence of phosphine ligands affords complexes of the formula, [M(NCOC2H4CO)(PPh3)2(Br)].13 The Pd-complex has proved to be an efficient and selective catalyst for Stille11 and Suzuki12 cross-coupling reactions, and the succinimidate ligand, a pseudohalide that has been described as a σ-acceptor14 and moderate π-donor (stronger π-donor than chlorine),15 plays an interesting role in the catalytic process. We have also synthesised bridged di-µ-imidate palladacyclopentadiene complexes starting from the appropriate binuclear hydroxo palladium complex [NBu4]2[Pd2{C4(COOMe)4}2(µ-OH)2],16 by means of a simple acid–base reaction, which is well established in our group.17 The hemilabile behaviour18 of the new di-µ-imidate complexes towards monodentate ligands, such as acetonitrile, phosphines (Ph3P, (p-C6H4F)3P and n-Bu3P), pyridine, p-toluidine and tetrahydrothiophene, has allowed the preparation of mononuclear imidate palladacyclopentadiene derivatives, which are catalytically active in the Stille reaction. A comparison of their catalytic properties with [1,2,3,4-tetrakis(methoxycarbonyl)-1,3-butadiene-1,4-diyl]palladium(II) (TCPC), Pd(NCOC2H4CO)(PPh3)2Br11b and Pd(NCOC2H4CO)(dppe)Br in the Stille reaction has been reported, revealing an interesting dependence in both the yields and reaction times on the presence and type of imidate ligand bound to Pd. We have also recently described the preparation of mono- and dinuclear imidato complexes with a classical C⁁N-cyclometallated backbone (C⁁N = phenylazophenyl (azb, a) or 2-pyridylphenyl (phpy, b)) employing, as precursors, the well known complexes with bridging acetate groups.19 The structural properties of mononuclear derivatives of the type, [Pd(azb)(succinimidate)(PR3)], were investigated in a subsequent study.20
In view of the successful catalytic results, vide supra, in relation to C⁁N–palladacyclic complexes and our experience in this field with other imidato complexes, we report here the preparation of new dinuclear orthometallated palladium(II) derivatives, possessing a 7,8-benzoquinolyl (bzq, c) ligand with bridging succinimidate, maleimidate or phthalimidate ligands; their use as precursors to mononuclear compounds of the type [Pd(C⁁N)(imidate)(PR3)]; and further the application of the novel complexes as catalysts for Suzuki–Miyaura coupling of aryl bromides with aryl boronic acids and the Sonogashira coupling reactions of aryl bromides with alkynes. The latter reaction has been studied in the presence and absence of a Cu(I) co-catalyst.
Results
Synthesis and characterisation
The acetato-bridged cyclometallated dimer [{Pd(µ-OOCMe)(bzq)}2] reacts with succinimide, phthalimide or maleimide in refluxing acetone to give dinuclear complexes in which the imidato ligands replace the bridging acetate group as presented in Scheme 1.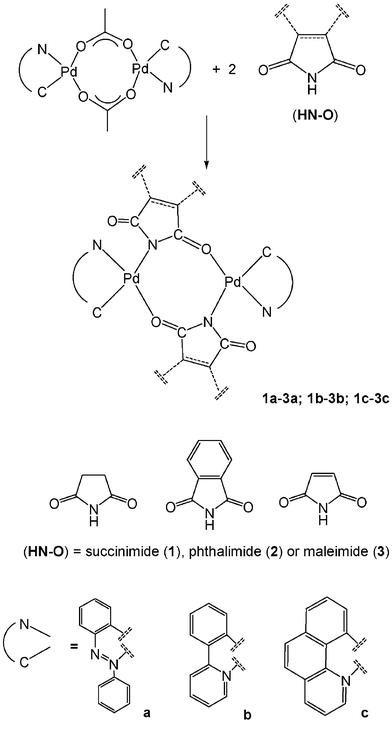 | ||
| Scheme 1 Synthesis of dinuclear complexes, 1a–3a, 1b–3b and 1c–3c. | ||
The new 7,8-benzoquinolyl derivatives are air-stable yellow solids and their infrared spectra show the characteristic absorptions of the cyclometallated ligand, partially overlapped with two strong bands attributed to imidato-carbonyl stretching, in the range 1736–1720 and 1570–1614 cm−1, respectively. According to previously reported data15b the appearance of two bands in this region suggests coordination of one carbonyl group to the metal as a part of an –NCO– bridging unit. Further evidence for the dinuclearity of complexes (1c–3c) comes from the FAB mass spectrometry, as inferred from the m/z values for the observed fragments. The abundances of the signals found around the parent ion are consistent with the expected natural isotopic distribution. We have also carried out TG, DTG, and DSC studies of dinuclear imidato complexes (1a–3a, 1b–3b, 1c–3c) in a dynamic nitrogen atmosphere. From this perspective, we have described previously21 the thermal analysis of mononuclear and halide-bridged ortho-palladated (azb and phpy based) complexes, and some studies dealing with the systematic thermal behaviour of cyclopalladated pseudohalogen-bridged complexes (dmba and bzan based) have been recently reported.22 The TG curves of our complexes display a one-stage decomposition pattern that reveals a high thermal stability, which is confirmed by the corresponding exothermic peaks in DSC (see Experimental section). A sharp mass loss is observed for succinimidate and phthalimidate derivatives, independent of the cyclometallated ligand, whereas maleimidate compounds 3a, 3b and 3c, show a slow and irregular decomposition. These data indicate the bzq and phpy derivatives exhibit similar and higher decomposition temperatures than the corresponding azb compounds.
Thermal stability is also influenced by the nature of the bridging ligand. Here the phthalimidate complexes are more stable as a series. The poor solubility of 1c–3c in common deuterated solvents, and the partial dissociation experienced in coordinating solvents such as CD3CN, thwarted detailed NMR spectroscopic analysis. However, the expected reaction of these imidato-bridged complexes (1c–3c), as well as the other bridged cyclometallated dimers (2a, 3a, 2b and 3b), with tertiary phosphines (PPh3, P(4-F-C6H4)3 and P(4-MeO-C6H4)3), gave the mononuclear N-bonded imidato complexes (Scheme 2, Table 1).
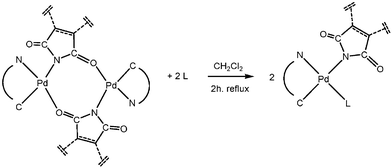 | ||
| Scheme 2 Synthesis of mononuclear imidate complexes. | ||
| C⁁N | Imidate | Ligand | Compound |
|---|---|---|---|
| azb = phenylazophenyl; phpy = 2-pyridylphenyl, bzq = 7,8-benzoquinolyl.a Complexes 4a–6a and 4b–6b have been previously described.19 | |||
| azb | Succinimidate | PPh3 | 4a a |
| azb | Succinimidate | P(4-F-C6H4)3 | 5a a |
| azb | Succinimidate | P(4-OMe-C6H4)3 | 6a a |
| azb | Phthalimidate | PPh3 | 7a |
| azb | Phthalimidate | — | 8a |
| azb | Phthalimidate | P(4-OMe-C6H4)3 | 9a |
| azb | Maleimidate | PPh3 | 10a |
| azb | Maleimidate | P(4-F-C6H4)3 | 11a |
| azb | Maleimidate | P(4-OMe-C6H4)3 | 12a |
| phpy | Succinimidate | PPh3 | 4b a |
| phpy | Succinimidate | P(4-F-C6H4)3 | 5b a |
| phpy | Succinimidate | P(4-OMe-C6H4)3 | 6b a |
| phpy | Phthalimidate | PPh3 | 7b |
| phpy | Phthalimidate | P(4-F-C6H4)3 | 8b |
| phpy | Phthalimidate | P(4-OMe-C6H4)3 | 9b |
| phpy | Maleimidate | PPh3 | 10b |
| phpy | Maleimidate | P(4-F-C6H4)3 | 11b |
| phpy | Maleimidate | P(4-OMe-C6H4)3 | 12b |
| bzq | Succinimidate | PPh3 | 4c |
| bzq | Succinimidate | P(4-F-C6H4)3 | 5c |
| bzq | Succinimidate | P(4-OMe-C6H4)3 | 6c |
| bzq | Phthalimidate | PPh3 | 7c |
| bzq | Phthalimidate | P(4-F-C6H4)3 | 8c |
| bzq | Phthalimidate | P(4-OMe-C6H4)3 | 9c |
| bzq | Maleimidate | PPh3 | 10c |
| bzq | Maleimidate | P(4-F-C6H4)3 | 11c |
| bzq | Maleimidate | P(4-OMe-C6H4)3 | 12c |
The reactions take place in refluxing CH2Cl2, yielding air stable solids with negligible molar conductivity. The IR spectra exhibit the expected bands for the corresponding cyclometallated backbone, together with those attributed to the incoming phosphine ligand and one strong carbonyl band around 1640 cm−1. The 1H NMR data of the mononuclear complexes is detailed in the Experimental section. These complexes exhibit the expected 1H resonances for cyclometallated and imidate ligands. The 31P NMR spectra of the complexes all show characteristic singlet resonances, with chemical shifts in the expected range for Pd(II) complexes (Table 2).
| Imidate | PPh3 | P(4-F-C6H4)3 | P(4-OMe-C6H4)3 | |
|---|---|---|---|---|
| Phenylazophenyl (azb) | phth | 41.5, 7a | 39.7, 8a | 38.7, 9a |
| mal | 41.9, 10a | 39.6, 11a | 38.6, 12a | |
| 2-Pyridylphenyl (phpy) | phth | 43.1, 7b | 40.9, 8b | 40.9, 9b |
| mal | 43.2, 10b | 40.9, 11b | 39.1, 12b | |
| 7,8-Benzoquinolyl (bzq) | succ | 42.9, 4c | 40.8, 5c | 39.3, 6c |
| phth | 43.0, 7c | 40.9, 8c | 39.5, 9c | |
| mal | 42.6, 10c | 40.9, 11c | 39.0, 12c |
Suitable crystals of 7c and 9a were grown from CH2Cl2 and diethyl ether, enabling their molecular structures to be confirmed by X-ray single crystal diffraction (Figs. 2 and 3, respectively, and Table 3). The deviation from the planar coordination has been quantified by the PCNPd and NNCPd improper torsion angles.19 The values are 2.66(3)° (P1C1N2Pd) and 0.28(5)° (N3N2C1Pd) in 9a, while they are 0.80(6)° (P1C1N1Pd) and 3.67(9)° (N2N1C1Pd) in 7c. Thus, the coordination around Pd is essentially planar in both complexes, the main distortion about the ideal geometry is the bite angle that involves the C⁁N ligand (Table 4).
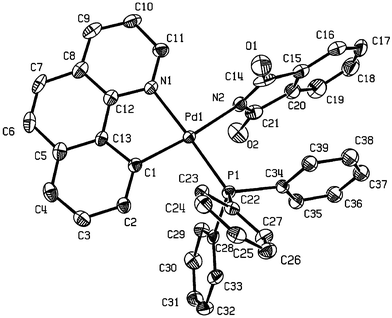 | ||
| Fig. 2 X-Ray crystal structure of 7c. Thermal elipsoids are at 50% probability. Hydrogens omitted for clarity. | ||
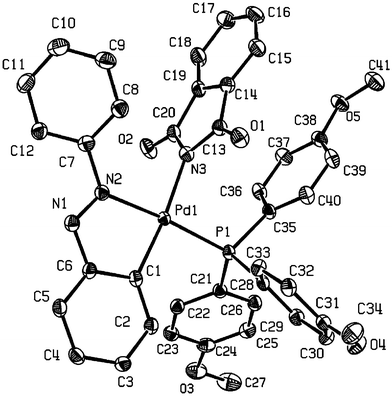 | ||
| Fig. 3 X-Ray crystal structure of 9a. Thermal elipsoids are at 50% probability. Hydrogens omitted for clarity. | ||
| 7c | 9a | |
|---|---|---|
| Empirical formula | C39H27N2O2PPd | C41H34N3O5PPd |
| Formula weight | 693.00 | 786.08 |
| T/K | 173(2) | 100(2) |
| λ/nm | 0.71073 | 0.71073 |
| Crystal system | Monoclinic | Triclinic |
| Space group | P21/c |
P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
| a/Å | 18.5176(16) | 9.2124(4) |
| b/Å | 10.5769(8) | 14.1362(7) |
| c/Å | 16.3681(13) | 14.3805(7) |
| α/° | 90 | 111.4360(10) |
| β/° | 111.731(7) | 94.2200(10) |
| γ/° | 90 | 94.7580(10) |
| V/Å3 | 2978.0(4) | 1726.26(14) |
| Z | 4 | 2 |
| ρ calc./Mg m−3 | 1.546 | 1.512 |
| μ/mm−1 | 0.717 | 0.635 |
| F(000) | 1408 | 804 |
| Crystal size/mm | 0.28 × 0.20 × 0.12 | 0.28 × 0.19 × 0.13 |
| Reflections collected | 7371 | 20063 |
| Independent reflections | 5236 | 7719 |
| Goodness-of-fit on F2 | 0.892 | 0.904 |
| Final R indices [I>2σ(I)] | R 1 = 0.0363, ωR2 = 0.0740 | R 1 = 0.0301, ωR2 = 0.0769 |
| R indices (all data) | R 1 = 0.0612, ωR2 = 0.0790 | R 1 = 0.0323, ωR2 = 0.0787 |
| 7c | 9a | |
|---|---|---|
| Pd(1)–P(1) | 2.2519(10) | 2.2768(15) |
| Pd(1)–C(1) | 2.022(3) | 2.0048(19) |
| Pd(1)–N(2) | 2.098(3) | 2.1088(16) |
| Pd(1)–N(1) | 2.098(3) | — |
| Pd(1)–N(3) | — | 2.0872(17) |
| C(1)–Pd(1)–N(3) | — | 173.80(7) |
| C(1)–Pd(1)–N(2) | 171.71(14) | — |
| C(1)–Pd(1)–N(2) | — | 78.08(7) |
| C(1)–Pd(1)–N(1) | 82.06(13) | — |
| N(3)–Pd(1)–N(2) | — | 95.73(6) |
| N(2)–Pd(1)–N(1) | 91.51(12) | — |
| C(1)–Pd(1)–P(1) | 95.35(11) | 95.69(6) |
| N(3)–Pd(1)–P(1) | — | 90.51(5) |
| N(2)–Pd(1)–P(1) | 91.16(9) | 172.78(5) |
| N(1)–Pd(1)–P(1) | 177.19(9) | — |
| N(1)–N(2) | — | 1.265(2) |
| C(6)–N(1) | — | 1.398(3) |
| C(7)–N(2) | — | 1.440(2) |
In both structures the imidate ligand is approximately perpendicular to the coordination plane, with angles between planes of 82.7(6)° and 81.7(10)° for 9a and 7c, respectively. Similar values are found in succinimidate complexes.20 Following the classification described by Dance and Scudder23 for PPh3, based on measures of the M–P–Cipso–C torsion angle, the conformations of Pd–PPh3 and Pd–P(4-MeO-C6H4)3 groups may be described as non-rotors. Here, there are two parallel phenyl rings and one orthogonal phenyl ring. The Pd–N2 bond length in 7c of 2.098(3) Å compares with a Pd–N bond length of 2.026(2) Å in trans-bis(phthalimide)Pd(PPh3)2,24 whereas the Pd–N3 bond length in 9a is 2.087(2) Å. Thus it appears that the phthalimidate ligand, in both 7c and 9a, is a weaker σ-acceptor, which is presumably associated with the greater electron-withdrawing potential of the Csp2⁁Nsp2 cyclometallated backbone in these complexes.
Application of the imidato-Pd-complexes as catalysts/ precatalysts in Suzuki–Miyaura cross-coupling
The Pd-complexes were screened for catalytic activity in the cross-coupling of three aryl bromides (13a–c) with phenyl boronic acid to give 14a–c, respectively (Scheme 3). Standard conditions, recently reported by us for other imidato-Pd(II) catalyst systems, were employed in these reactions. | ||
| Scheme 3 Benchmark Suzuki–Miyaura cross-coupling reactions. | ||
With a large number of Pd complexes to be screened, efforts were focused on use of the 7,8-benzoquinolyl (bzq) Pd-imidato series (1c–12c) and selected others, taken from the phenylazobenzene (azb) and 2-pyridylphenyl (phpy) series (7a, 7b, 8b, 9b, 10a, 10b, 11b and 12b). The catalyst loading used for the mononuclear complexes was 1 mol%, with an exception made for the dimeric complexes (1c–3c), where 0.5 mol% of each precursor was employed. Percentage conversion to products were recorded after 2 h for reactions employing 13a and 13b as substrates, and after 4 h for the reaction employing 13c — a less reactive substrate (10 mol% hexadecane was used as the internal standard). Multiple reactions were simultaneously quenched after identical reaction times to give an insight into the catalytic activity of each Pd complex, and thus their relative reactivity with these substrates.
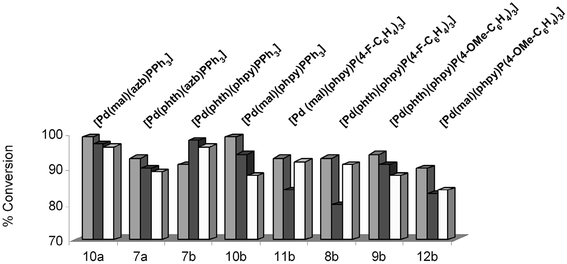
The dimeric cyclometallated precursors (1c–3c) all demonstrated reasonable catalytic activity in the three benchmark reactions, with yields seen between ca. 70 and 84% (Fig. 4). Given that these Pd sources do not possess a phosphine ligand, the catalytic activity and yields are promising. These yields are, however, improved by the incorporation of a phosphine ligand. In the reaction of 13a→14a, yields of >87% are seen for complexes 4c–6c, containing a succinimidate ligand. The best yield is seen for 6c (99%). In this sense, it appears that the more electron rich P(4-OMe-C6H4)3 ligand increases the reactivity of the Pd catalyst in 6c. This trend is also apparent in the other reactions (13b→14b and 13c→14c).
![Suzuki–Miyaura cross-coupling using Pd complexes 1c–12c in a THF/2M Na2CO3 mixture (1 ∶ 2, v/v, 3 mL total volume). Key: reaction of 13a→14a; ■ reaction of 13b→4b; □ reaction of 13c→14c. T
= 90 °C. Reaction time = 2 h for 13a→14a and 13b→14b, = 4 h for 13c→14c. Concentration of aryl halide [13]
= 0.15 M (1.1 equiv.). Concentration of phenyl boronic acid = 0.136 M (1.0 equiv.). % Conversion was determined by GC analysis using hexadecane as an internal standard.](/image/article/2004/DT/b413886d/b413886d-f4.gif) | ||
Fig. 4 Suzuki–Miyaura cross-coupling using Pd complexes 1c–12c in a THF/2M Na2CO3 mixture (1 ∶ 2, v/v, 3 mL total volume). Key: ![[square tinted (heavy)]](https://www.rsc.org/images/entities/char_e069.gif) reaction of 13a→14a; ■ reaction of 13b→4b; □ reaction of 13c→14c. T
= 90 °C. Reaction time = 2 h for 13a→14a and 13b→14b, = 4 h for 13c→14c. Concentration of aryl halide [13]
= 0.15 M (1.1 equiv.). Concentration of phenyl boronic acid = 0.136 M (1.0 equiv.). % Conversion was determined by GC analysis using hexadecane as an internal standard. reaction of 13a→14a; ■ reaction of 13b→4b; □ reaction of 13c→14c. T
= 90 °C. Reaction time = 2 h for 13a→14a and 13b→14b, = 4 h for 13c→14c. Concentration of aryl halide [13]
= 0.15 M (1.1 equiv.). Concentration of phenyl boronic acid = 0.136 M (1.0 equiv.). % Conversion was determined by GC analysis using hexadecane as an internal standard. | ||
Some trends are apparent upon altering the type of imidate ligand among Pd complexes 5c, 8c and 11c, which all contain P(4-F-C6H4)3 as the phosphine ligand. For example, analysis of the reaction 13b→14b reveals an interesting difference. We observe a 90% conversion to 14b for 5c (containing succinimidate), 91% for 8c (containing phthalimidate) and 83% for 11c (containing maleimidate). We believe the greatest difference, of 8% conversion seen between 8c and 11c, is real. In reactions that are run in parallel, under identical conditions with multiple injections (stirring rate, temperature, reagent, solvent and base sources), we estimate that the conversion error to cross-coupled products is ± 3% (Note: greater errors, ± 5%, are observed on performing reactions separately). Hence the choice of imidate, the anionic ligand, does influence the overall conversion to products. However this effect is also dependent upon the particular phosphine ligand, and a subtle interplay of electron-donation and withdrawal could account for these differences. These effects may also be substrate specific.
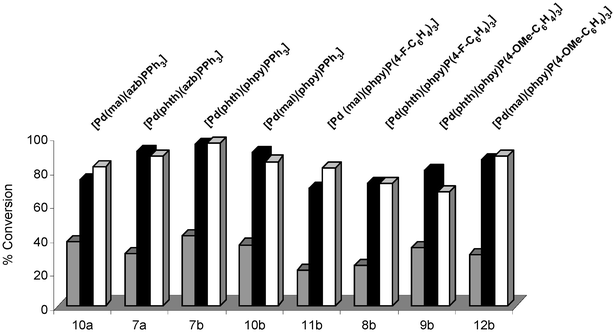
We have further noted that a difference in catalytic activity is seen on changing the type of cyclometallated backbone (Fig. 5). For example, taking the reaction of 13c→14c, and comparing Pd complexes containing malelimidate and PPh3 ligands (10a–c), a notable difference (≈10%) is seen between the 2-pyridylphenyl backbone (10b, 87%) and both the phenylazabenzene (10a, 97%) and 7,8-benzoquinolyl backbones (10c, 98%).
 | ||
| Fig. 5 As for Fig. 4, using Pd complexes 7a, 7b, 8b, 9b, 10a, 10b, 11b and 12b. | ||
It is not unexpected that individual changes in the catalyst structure affect the overall product conversions. Presumably the cyclometallated backbone will directly affect the rate of catalyst initiation, assuming a classic Pd(II)→Pd(0) pre-reduction step, and thus the concentration of catalytically active ‘Pd’. The outcome here is that the cyclometallated backbone, type of imidate and presence of phosphine ligand all affect this particular cross-coupling process.
Application of the imidato-Pd-complexes as catalysts/ precatalysts in Sonogashira cross-coupling
To gain further insight into the influence of the various ligands, we decided to apply our Pd-complexes to the Sonogashira cross-coupling25 of 4-bromoacetophenone 13c and phenyl acetylene to give 15 (Scheme 4). Although, 13c is an activated substrate, the reaction is not as straight-forward as one might first assume. When the reaction is mediated by (PPh3)2PdCl2 (1 mol%) in the presence of CuI (0.5 mol%) in an acetonitrile/triethylamine mixture (2.5 ∶ 1.5, v:v), a reaction temperature of 100 °C is required to achieve even 63% percent conversion after 2 h (c of 13c = 0.102 M). | ||
| Scheme 4 Benchmark Sonogashira cross-coupling reaction. | ||
It should be noted that the reactant concentration influences the rate considerably, and in due course we will present our findings regarding this generic precatalyst. One point to note here is that, as expected, higher concentrations give higher reaction rates, but favour side-products such as the homo-coupled phenyl acetylene dimer (1,4-bis(phenylethynyl)buta-1,3-diyne). In these reactions, we did not detect hydrodebrominated acetophenone, which would facilitate reoxidation of Pd(0) to Pd(II) to promote homo-coupling.26 It is presumed, although not proven, that adventitious oxygen promotes the homo-coupling process at higher concentrations. Keeping the concentration of 13c <0.13 M serves to minimise this to the level expected from the Pd(II)→Pd(0) pre-reduction step. For screening purposes, we employed the general conditions described above for (PPh3)2PdCl2 (Figs. 6 and 7). We have further performed the reactions under ‘copper-free’27 conditions to assess the impact of Cu(I) on the rate of reaction and on the production of side-products.
![Sonogashira cross-coupling of 4-bromoacetophenone 13c with phenylacetylene to give 15 in a Et3N/CH3CN mixture (2 ∶ 3, v/v, 5 mL total volume). Key:
■ in the absence of CuI, % conv. after 2 h; in the absence of CuI, % conv. after 5 h; □ in the presence of CuI, % conv. after 2 h. T
= 100 °C. Concentration of aryl halide [13c]
= 0.102 M (1.04 equiv.). Concentration of phenyl acetylene = 0.098 M (1.0 equiv.). % Conversion was determined by GC analysis using hexadecane as an internal standard.](/image/article/2004/DT/b413886d/b413886d-f6.gif) | ||
| Fig. 6 Sonogashira cross-coupling of 4-bromoacetophenone 13c with phenylacetylene to give 15 in a Et3N/CH3CN mixture (2 ∶ 3, v/v, 5 mL total volume). Key:
■ in the absence of CuI, % conv. after 2 h; in the absence of CuI, % conv. after 5 h; □ in the presence of CuI, % conv. after 2 h. T
= 100 °C. Concentration of aryl halide [13c]
= 0.102 M (1.04 equiv.). Concentration of phenyl acetylene = 0.098 M (1.0 equiv.). % Conversion was determined by GC analysis using hexadecane as an internal standard. | ||
 | ||
| Fig. 7 As for Fig. 6, using Pd complexes 7a, 7b, 8b, 9b, 10a, 10b, 11b and 12b. | ||
The first major observation that comes out of screening complexes 1c–12c is that the presence of CuI serves to accelerate the reaction across the whole series. Thus for these specific Pd-catalysts/precatalysts, the Cu(I) salt serves to enhance the rate of reaction and not to inhibit it, as reported in a unique case by Buchwald and co-workers.27e In the absence of CuI, reactions take 5 h to reach similar conversions achievable in only 2 h in the presence of CuI. Although the copper-free Sonogashira reactions did require longer reactions times, we found that the GC spectra were cleaner in a number of the examples illustrated in Figs. 6 and 7. As was the case with the Suzuki–Miyaura cross-coupling reactions, the presence of a phosphine ligand serves to improve the overall conversion to 15 (compare 1c–3c with 4c–12c).
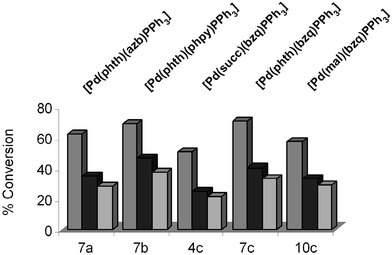
In the presence of CuI, the type of imidate again affects the overall conversion for complexes containing the P(4-F-C6H4)3 ligand and 7,8-benzoquinolyl backbone (5c, 8c and 11c, Fig. 6). The highest conversion is seen for 5c containing a succimidate ligand (86%), whereas alteration to a phthalimidate ligand (8c) results in a drop to 76%. The lowest conversion is seen in 11c, possessing a maleimidate ligand (69%). The difference between 5c and 11c of 17% is notable. Although such large differences are not seen for all the phosphine ligands, clearly the imidate plays a large role in either catalyst initiation or in the catalytic cycle.
| [Pd] Catalyst | 0.1 mol% [Pd] | 0.01 mol% [Pd] | 0.001 mol% [Pd] | ||||||
|---|---|---|---|---|---|---|---|---|---|
| % Conv. | TON | TOF | % Conv. | TON | TOF | % Conv. | TON | TOF | |
| 7a | 62 | 620 | 103 | 35 | 3500 | 583 | 28 | 28000 | 4666 |
| 7b | 69 | 690 | 115 | 46 | 4600 | 766 | 37 | 37000 | 6166 |
| 4c | 50 | 500 | 83 | 25 | 2500 | 416 | 21 | 21000 | 3500 |
| 7c | 70 | 700 | 116 | 40 | 4000 | 666 | 33 | 33000 | 5500 |
| 10c | 57 | 570 | 95 | 33 | 3300 | 550 | 29 | 29000 | 4833 |
 | ||
| Fig. 8 Sonogashira cross-coupling, in the absence of CuI, of 4-bromoacetophenone 13c with phenylacetylene to give 15 at various Pd catalyst loadings. Reaction time = 6 h, T
= 100 °C. Reactant concentrations as for Fig. 6. Key:
(0.1 mol% Pd); ■
(0.01 mol% Pd);
(0.001 mol% Pd).% Conversion was determined by GC analysis using hexadecane as an internal standard. | ||
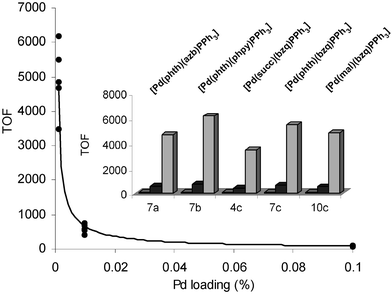 | ||
| Fig. 9 Graph to show turnover frequency versus Pd loading (TOF = (mol of coupled product) (mol Pd)−1 h−1). Insert graph is as for Fig. 8. | ||
The highest TOF and TON were seen using 7b at a loading of 0.001 mol% (TOF = 6166; TON = 37,000). The difference in the type of cyclometallated backbone is apparent once again (compare 7b with 7a (TOF = 4666; TON = 28,000) and 7c (TOF = 5500; TON = 33,000), containing 2-pyridylphenyl, phenylazobenzene and 7,8-benzoquinolyl backbones, respectively). A comparison of 4c, 7c and 10c allows us to directly explore the influence of the imidate ligand. The phthalimidate ligand in 7c is clearly more active, in order, than 10c (TOF = 4833; TON = 29,000), containing a maleimidate ligand, and 4c, containing a succinimdate ligand (TOF = 3500; TON = 21,000). The reaction profiles for 4c, 7c and 10c showed the initial rates and associated TOF differences were essentially constant throughout the reaction.
Catalyst recycling in Suzuki–Miyaura cross-coupling
It is well known that one of the major drawbacks of homogeneous palladium catalysed cross-coupling reactions is product separation from the catalyst (phosphine ligands, and their oxides, can further complicate purification.30 Lower catalyst loadings usually give purer cross-coupled products, however these active catalysts are often even more difficult to recover for recycling. Given that loadings of 1 mol% were used in our Suzuki–Miyarua cross-coupling reactions, vide supra, we decided to investigate whether it was possible to recycle these catalysts. Our method for catalyst recycling is based on a simple protocol developed by Monteiro and co-workers for Pd(OAc)2/PPh3.31 They demonstrated that a poly(ethylene oxide)32 (PEO)/methanol solvent medium can be used in Suzuki–Miyaura cross coupling. At the end of the reaction, the product was extracted into a non-polar phase, with the Pd-catalyst remaining in the polar phase. Recharging the polar phase with new reactants allows the Pd to be re-used without loss of catalytic activity (over several runs). Two reactions were chosen for the catalyst recycling studies, namely reaction 13a→14a, and 13c→14c. Complexes 4c and 7c were evaluated for recycling and the results are shown in Figs. 10 and 11.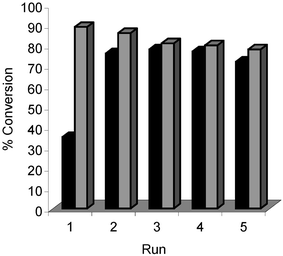 | ||
| Fig. 10 Reaction of 4-bromonitrobenzene 13a with phenyl boronic acid to give 14a. ■ Complex 4c; Complex 7c. T
= 90 °C. Reaction time = 2 h. | ||
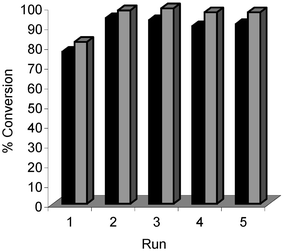 | ||
| Fig. 11 Reaction of 4-bromoanisole 13c with phenyl boronic acid to give 14c. ■ Catalyst 4c; Catalyst 7c. T
= 90 °C. Reaction time = 4 h. | ||
In both reactions, 13a→14a and 13c→14c, efficient catalyst recycling is demonstrated, without appreciable loss of catalytic activity over five consecutive catalytic runs. Complex 7c is more active than 4c in both reactions, throughout the reaction series.
Discussion and conclusions
Influence of copper in Sonogashira cross-coupling
It is clear that Cu(I) serves as a co-catalyst and accelerates the rate of Sonogashira cross-coupling in the presence of the cyclometalled Pd(II) precursors. However, a closer examination of Figs. 6 and 7 reveals that the magnitude of this enhancement depends upon the precise structure of the Pd(II) precatalyst. Consideration of the binuclear imidate bridged Pd(II) precatalysts (1c–3c), which possess a common 7,8-benzoquinolinyl backbone, finds that Cu(I) is not equally efficacious across the range of imidates. Thus copper promotional magnitude appears related to the ability of each imidate to delocalise electron density from the nitrogen lone pair on to the carbonyl oxygen. Hence, delocalising ability follows the order, maleimide (3c) > phthalimide (2c) > succinimide (1c), while the Cu(I) rate-enhancement follows the reverse sequence (Fig. 12).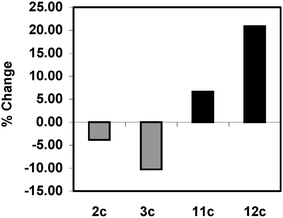 | ||
| Fig. 12 Relative effect of added Cu(I) salt for different imidate and phosphine ligands:
% change for binuclear complexes 2c and 3c relative to 1c; ■
% change for mononuclear complexes 11c and 12c relative to 10c. | ||
For mononuclear cyclometallated Pd(II) precatalysts, the influence of Cu(I) on the reaction was also dependent on the choice of phosphine. For the series of maleimidate based complexes, possessing the same 7,8-benzoquinolinyl backbone (10c–12c), the benefit of Cu(I) promotion scaled with the electron-donor properties of the phosphine ligand (Fig. 12). Here the strongly-donating methoxy substituent (12c) works in synergy with copper giving the greatest enhancement, followed by the fluorene substituent (11c), which in turn is superior to Ph3P (10c).
Recycling studies
Two of our palladium precatalysts (4c and 7c) can be successfully supported and recycled in Suzuki–Miyaura cross-coupling reactions. In both reactions (13a→14a and 13c→14c), 7c was always superior to 4c. Catalyst 4c also exhibited an induction/activation phase as apparent from the low conversions during the first runs, which may be linked to its intrinsically poorer performance. The importance of precatalyst activation is illustrated by our finding that, provided an initial run was conducted at 90 °C, all subsequent reactions using recycled catalysts could be performed at temperatures as low as 25 °C without loss in activity. The thermal stability of the initial precatalyst complexes clearly impacts on these reactions.Summary of structural effects — imidate, cyclometallated backbone and phosphine
The body of results collected herein demonstrates that all parts of the ligand framework play a role in the cross-coupling reactions evaluated. The choice of imidate is clearly a major factor as illustrated for the reaction 13c→15 wherein succinimidate 5c, phthalimidate 8c and maleimidate 11c, provide conversions of 86%, 76% and 69% respectively. Phosphine electronics are also important in both Sonogashira and Suzuki–Miyaura couplings, affecting conversions by as much as ± 20% as shown in Fig. 12. Although changes to the cyclometallated backbone have some effect on Sonogashira chemistry, their influence is somewhat less than that exerted by the anionic imidate and phosphine ligands, as evidenced by the similar conversions for 7a–c seen in Fig. 8. However, it is difficult to deconstruct the complex, interdependent interactions within the ligand framework which undoubtedly control the formation, propagation and stability of important Pd species within the catalytic cycle.Influence of Pd loading in copper-free Sonogashira cross coupling
We have uncovered the first evidence that catalyst loading is crucial in regulating Pd-catalysed Sonogashira cross-couplings. In the reaction of 13c with phenyl acetylene to give 15, the TOFs were approximately inversely proportional to the Pd loading, which were highest at 0.001 mol%. We anticipate that higher loadings lead to aggregation of the various palladium species that are present in the reaction medium. It would also be interesting to study the effect of Cu(I) salts in this case, to see whether a similar trend was observed.Experimental
General details
THF was dried over sodium–benzophenone ketyl (distilled prior to use). All reactions were conducted under an inert atmosphere of Ar or N2 on a Schlenk line. Melting points were recorded on an electrothermal IA9000 Digital Melting Point Apparatus and are uncorrected. TLC analysis was performed on Merck 5554 aluminium backed silica gel plates and compounds visualized by ultraviolet light (254 nm), phosphomolybdic acid solution (5% in EtOH), or 1% ninhydrin in EtOH. 1H NMR spectra were recorded at 270 MHz using a JEOL EX270 spectrometer or at 400 MHz using a JEOL ECX400 spectrometer; 13C NMR spectra at 67.9 or 100.5 MHz. Chemical shifts are reported in parts per million (δ) downfield from an internal tetramethylsilane reference. Coupling constants (J values) are reported in hertz (Hz), and spin multiplicities are indicated by the following symbols: s (singlet), d (doublet), t (triplet), q (quartet), qn (quintet), sx (sextet), m (multiplet), br (broad). C, H, and N analyses were carried out with a Carlo Erba instrument. IR spectra were recorded on a Perkin-Elmer spectrophotometer 16F PC FT-IR, using Nujol mulls between polyethylene sheets. NMR data (1H and 31P) were recorded on Bruker Avance 200, 300 and 400 spectrometers. Mass spectrometric analyses were performed on a Fisons VG Autospec double-focusing spectrometer, operated in positive mode. Ions were produced by fast atom bombardment (FAB) with a beam of 25-keV Cs atoms. The mass spectrometer was operated with an accelerating voltage of 8 kV and a resolution of at least 1000. Conductance measurements were performed with a Crison 525 conductimeter (in acetone solutions, 5 × 10−4M). TG and DTG curves were recorded on a Mettler TG-50 thermobalance using an atmosphere of pure nitrogen (50 cm3 min−1; heating rate 5 °C min−1). DSC curves were recorded on a DSC822e Mettler Toledo instrument. All the solvents were dried by conventional methods.The cyclometallated precursor [{Pd(µ-OOCMe)(bzq)}2] [bzq = 7,8-benzoquinolyl] was synthesized following the reported method.33 Dinuclear imidato precursors [{Pd(µ-NCO)(C⁁N)}2] [C⁁N = phenylazophenyl (azb); –NCO– = succinimidate (suc) (1a), phthalimidate (2a) (phth) or maleimidate (mal) (3a); C⁁N = 2-pyridylphenyl (phpy); –NCO– = succinimidate (1b), phthalimidate (2b) or maleimidate (3b)] and mononuclear succinimidate derivatives [Pd(C⁁N)(suc)(L)] [C⁁N = azb; L = PPh3 (4a), P(4-F-C6H4)3 (5a), P(4-MeO-C6H4)3 (6a); C⁁N = phpy; L = PPh3 (4b), P(4-F-C6H4)3 (5b), P(4-MeO-C6H4)3 (6b)] were prepared as described in a previous paper.18 Commercially available chemicals were purchased from Aldrich Chemical Co. and were used without further purification.
Preparation of the complexes [{Pd(µ-NCO)(bzq)}2] [bzq = 7,8-benzoquinolyl; –NCO– = succinimide (succ) (1c), phthalimide (2c) (phth) or maleimide (mal) (3c)]
The new complexes were obtained by treating [{Pd(µ-OOCMe)(bzq)}2] with the corresponding imide (molar ratio 1 ∶ 2) in acetone, according to the following general method. To an acetone (20 mL) solution of [{Pd(µ-OOCMe)(bzq)}2] (0.175 g, 0.255 mmol) was added the stoichiometric amount of ligand. The resulting suspension was refluxed for 2 h, and then concentrated to one fifth of the initial volume. Slow addition of diethyl ether completed the precipitation of the title complexes, which were isolated by filtration, washed with diethyl ether and air-dried.Thermal analysis of dinuclear complexes 1a–3a and 1b–3b
Preparation of complexes [Pd(C⁁N)(imidate)(L)] [C⁁N= azb; imidate = phth; L = PPh3 (7a), P(4-F-C6H4)3 (8a), P(4-MeO-C6H4)3 (9a); C⁁N= azb; imidate = mal; L = PPh3 (10a), P(4-F-C6H4)3 (11a), P(4-MeO-C6H4)3 (12a); C⁁N = phpy; imidate = phth; L = PPh3 (7b), P(4-F-C6H4)3 (8b), P(4-MeO-C6H4)3 (9b)); C⁁N = phpy; imidate = mal; L = PPh3 (10b), P(4-F-C6H4)3 (11b), P(4-MeO-C6H4)3 (12b); C⁁N= bzq; imidate = succ; L = PPh3 (4c), P(4-F-C6H4)3 (5c), P(4-MeO-C6H4)3 (6c); C⁁N= bzq; imidate = phth; L = PPh3 (7c), P(4-F-C6H4)3 (8c), P(4-MeO-C6H4)3 (9c); C⁁N= bzq; imidate = mal; L = PPh3 (10c), P(4-F-C6H4)3 (11c), P(4-MeO-C6H4)3 (12c)]
The complexes were obtained by treating the appropriated precursor [{Pd(µ-NCO)(C⁁N)}2] with the corresponding neutral ligand (molar ratio 1 ∶ 2) in dichloromethane, according to the following general method. To a dichloromethane (20 mL) solution of (2a, 3a, 2b, 3b or 1c–3c) (0.07 g) was added the stoichiometric amount of phosphine ligand. The solution was refluxed for 2 h, then concentrated until ca. one fifth of the initial volume. Slow addition of hexane caused the precipitation of the title complexes, which were isolated by filtration, washed with hexane, air-dried and recrystallised from acetone–hexane.Crystal structure determination of [Pd(azb)(phth)(P(4-MeO-C6H4)3)] 9a and [Pd(bzq)(phth)(PPh3)] 7c
Crystals suitable for a diffraction study were grown from dichloromethane/diethyl ether (complexes 7c and 9a). Data collection for 9a was performed at 100 K on a Bruker Smart CCD diffractometer with a nominal crystal to detector distance of 6.2 cm, diffraction data were collected based on a ω scan run. A total of 2524 (9a) were collected at 0.3° intervals and 10 s frame−1. The diffraction frames were integrated using the SAINT package34 and corrected for absorption with SADABS.35 Data collection for 7c was performed at 173 K on a Siemens P4 diffractometer. The structures were solved by Patterson (9a) and direct (7c) methods36 and refined by full-matrix least-squares techniques using anisotropic thermal parameters for non-H atoms 23 (Table 3).CCDC reference numbers 249864 and 249865.
See http://www.rsc.org/suppdata/dt/b4/b413886d/ for crystallographic data in CIF or other electronic format.
Typical Suzuki–Miyaura cross-coupling reaction
All reactions were performed in a Radleys carousel adapted for rigorous inert atmosphere reactions. The aryl boronic acid (0.41 mmol), aryl halide (0.45 mmol), Na2CO3 (1 M (aq.), 1 mL), THF (1.5 mL), hexadecane (0.1 equiv., internal standard) and Pd catalyst (0.01 equiv.) were degassed via ‘freeze–pump–thaw’ cycles (on a vacuum line, then transferred to the carousel). The resulting mixture was heated at 90 °C for the specified time and then quenched by addition of water (10 mL). The mixture was extracted with CH2Cl2 (3 × 10 mL), which was combined and washed with saturated NaCl (aq.) (1 × 10 mL), dried (MgSO4), filtered and then concentrated in vacuo.Typical Sonogashira cross-coupling reaction
All reactions were performed in a Radleys carousel adapted for rigorous inert atmosphere reactions. To the aryl halide 13c (0.51 mmol, 1.04 equiv.), hexadecane (0.1 equiv., internal standard) and Pd catalyst (X mol%; X = 1, 0.1, 0.01 or 0.001) was added dry degassed CH3CN (3.0 mL) and Et3N (2.0 mL). Phenylacetylene (0.49 mmol, 1.0 equiv.) and CuI (0.05 equiv.) were added (the latter was absent in the copper-free experiments). The reaction was heated to 100 °C and samples (250 µL) were withdrawn at regular time points. Each sample was immediately flushed through a plug of silica (0.65 g; Kieselgel 60, particle size 0.035–0.070 mm, 220–440 mesh, purchased from Fluka) packed in a Pasteur pipette with CH2Cl2 (2.5 mL). The resultant fraction was then treated with 1,2-bis(diphenylphosphino)ethane (dppe), at a final concentration containing four equivalents of dppe per maximum theoretical amount of palladium. This latter preparative sampling step ensures that no further cross-coupling occurs at room temperature (the time dependence of post-quenched samples showed no further changes using this method).37 The sample was then analysed by HRGC analysis.Typical procedure for catalyst recycling in Suzuki-Miyaura cross-coupling reaction
Using identical substrate quantities to those reported, but using an adapted procedure: all reactions/runs were carried out in a Radleys carousel adapted for rigorous inert atmosphere reactions. To an oven-dried carousel tube was added the Pd catalyst (0.01 equiv.), K3PO4 (0.5 mmol, 2 equiv.), PEO (1 g, Mw = 100,000) and phenylboronic acid (0.375 mmol, 1.5 equiv.). The flask was evacuated and back filled with N2 and then the aryl halide (0.25 mmol, 1 equiv.) in dry methanol (4 mL) was added. The reaction mixture was stirred at 90 °C for the desired time (as specified in the text). The cross-coupled product was extracted under a stream of N2 with hexane/diethyl ether (9 ∶ 1, v/v) (4 mL ca. four extractions). The combined apolar phase was washed with aqueous NaOH (1 M, 5 mL), saturated NaCl (aq.) (2.5 mL), and then dried (MgSO4). Concentration in vacuo afforded the biaryl product in > 90% purity as judged by 1H NMR spectroscopy and HRGC. For the recycle reactions, the polar phase was transferred to a an oven-dried carousel tube under N2. Then, the aryl halide (0.25 mmol), phenylboronic acid (0.375 mmol) and K3PO4 (0.5 mmol) were added. The reaction mixture was stirred at 90 °C for the desired time and the product was isolated as described above (note: that the reactions become very difficult to stir after the third/forth runs, however this does not appear to affect the yields of the cross-coupled products).Acknowledgements
I. J. S. F. thanks for the University of York for financial assistance (innovation and research priming fund grant) and Johnson Matthey PLC (UK) for a generous loan of palladium salts. A. F. L. thanks the University of York for laboratory refurbishment. Financial support of this work by Dirección General de Investigación (coordinated project-BQU2001-0979-C02-01/02) is gratefully acknowledged. The referees are thanked for their constructive comments.References and notes
- (a) Metal-catalyzed Cross-coupling Reactions, ed. F. Diederich and P. J. Stang, Wiley-VCH, New York, 1998 Search PubMed; (b) J. Tsuji, Palladium Reagents and Catalysts, Innovations in Organic Synthesis, Wiley, New York, 1995 Search PubMed.
- For a review on the Suzuki reaction, see: (a) A. Suzuki, J. Organomet. Chem,., 1999, 576, 147–168 CrossRef CAS; (b) A. Suzuki, Handbook of Organopalladium Chemistry for Organic Synthesis, John Wiley & Sons, 2002, pp. 249–262 Search PubMed.
- K. C. Nicolaou and E. J. Sorensen, Classics in Total Synthesis, VCH, New York, 1996, ch. 31 Search PubMed.
- Recent developments in Heck type reactions have been reviewed: (a) W. A. Herrmann, V. P. W. Böhm and C-P. Reisinger, J. Organomet. Chem., 1999, 576, 23–41 CrossRef CAS; (b) A. F. Littke and G. C. Fu, Angew. Chem. Int. Ed., 2002, 41, 4176–4211 CrossRef CAS; (c) W. A. Herrmann, K. O. D. Preysing and S. K. Schneider, J. Organomet. Chem., 2003, 687, 229–248 CrossRef CAS; (d) R. B. Bedford, Chem. Commun., 2003, 1787–1796 RSC.
- J. Dupont, M. Pfeffer and J. Spencer, Eur. J. Inorg. Chem., 2001, 1917–1927 CrossRef CAS.
- D. A. Albisson, R. B. Bedford and P. N. Scully, Tetrahedron Lett., 1998, 39, 9793–9796 CrossRef.
- (a) S. Iyer and C. Ramesh, Tetrahedron Lett., 2000, 41, 8981–8984 CrossRef CAS; (b) P. Beletskaya, A. N. Kashin, N. B. Karlstedt, A. V. Mitin, A. V. Cheprakov and G. M. Kazankov, J. Organomet. Chem., 2001, 622, 89–96 CrossRef CAS; (c) F. Yang, Y. Zhang, R. Zheng, J. Tie and M. He, J. Organomet. Chem., 2002, 651, 146–148 CrossRef CAS.
- H. Weissman and D. Milstein, Chem. Commun., 1999, 1901–1902 RSC.
- (a) D. Alonso, C. Nájera and M. C. Pacheco, J. Org. Chem., 2002, 67, 5588–5594 CrossRef CAS; (b) L. Botella and C. Nájera, Angew. Chem., Int. Ed., 2002, 41, 179–181 CrossRef CAS.
- (a) R. B. Bedford, C. S. J. Cazin, S. J. Coles, T. Gelbrich, P. N. Horton, M. B. Hursthouse and M. E. Light, Organometallics, 2003, 22, 987–999 CrossRef CAS; (b) R. B. Bedford, C. S. J. Cazin, S. J. Coles, T. Gelbrich, M. B. Hursthouse and V. J. M. Scordia, Dalton Trans., 2003, 3350–3356 RSC.
- C. M. Crawforth, I. J. S. Fairlamb and R. J. K. Taylor, Tetrahedron Lett., 2004, 45, 461–465 CrossRef CAS.
- I. J. S. Fairlamb, A. R. Kapdi, J. M. Lynam, R. J. K. Taylor and A. C. Whitwood, Tetrahedron, 2004, 60, 5711–5718 CrossRef CAS.
- (a) J. L. Serrano, Y. Zheng, J. R. Dilworth and G. Sánchez, Inorg. Chem. Commun., 1999, 2, 407–410 CrossRef CAS; (b) C. M. Crawforth, S. Burling, I. J. S. Fairlamb, R. J. K. Taylor and A. C. Whitwood, Chem. Commun., 2003, 2194–2195 RSC.
- It is excepted that N-donors are generally strong σ-donors, and that this effect will generally dominate σ-acceptor effects through bonds to the two neighbouring carbonyl groups.
- (a) D. M. Roundhill, Inorg. Chem., 1970, 9, 254–258 CrossRef CAS; (b) H. Adams, N. Bailey, T. N. Briggs, J. A. McCleverty, H. M. Colquhoun and D. J. Williams, J. Chem. Soc., Dalton Trans., 1986, 813–819 RSC.
- J. L. Serrano, I. J. S. Fairlamb, G. Sánchez, L. García, J. Pérez, J. Vives, G. López, C. M. Crawforth and R. J. K. Taylor, Eur. J. Inorg. Chem., 2004, 2706–2715 CrossRef CAS.
- (a) J. Ruiz, V. Rodriguez, N. Cutillas, M. Pardo, J. Pérez, G. López, P. A. Chaloner and P. B. Hitchcock, Organometallics, 2001, 20, 1973–1982 CrossRef CAS; (b) J. Ruiz, V. Rodríguez, G. López, J. Casabó, E. Molins and C. Miravitlles, Organometallics, 1999, 18, 1177–1184 CrossRef CAS and references cited therein; (c) J. Ruiz, N. Cutillas, V. Rodríguez, J. Sampedro, G. López, P. A. Chalonner and P. B. Hitchcock, J. Chem. Soc., Dalton Trans., 1999, 2939–2946 RSC; (d) G. Sánchez, J. L. Serrano, J. García, G. López, J. Pérez and E. Molins, Inorg. Chim. Acta, 1999, 287, 37–46 CrossRef CAS; (e) G. Sánchez, J. L. Serrano, J. Pérez, M. C. Ramírez de Arellano, G. López and E. Molins, Inorg. Chim. Acta, 1999, 295, 136–145 CrossRef CAS.
- (a) P. Braunstein and F. Naud, Angew. Chem. Int. Ed., 2001, 40, 680–699 CrossRef CAS; (b) C. S. Slone, D. A. Weinberger and C. A. Mirkin, Prog. Inorg. Chem., 1999, 48, 233–250 CAS.
- J. L. Serrano, L. García, J. Pérez, E. Pérez, J. Vives, G. Sánchez, G. López, E. Molins and A. Guy Orpen, Polyhedron, 2002, 21, 1589–1596 CrossRef CAS.
- J. Pérez, L. García, E. Pérez, J. L. Serrano, J. F. Martínez, G. Sánchez, G. López, A. Espinosa, M. Liu and F. Sanz, New J. Chem., 2003, 27, 1490–1496 RSC.
- (a) J. Pérez, G. Sánchez, J. García, J. L. Serrano and G. López, Thermochim. Acta, 2000, 362, 59–70 CrossRef CAS; (b) J. Pérez, G. Sánchez, J. García, J. L. Serrano and G. López, J. Therm. Anal. Cal., 2001, 66, 361–370 Search PubMed.
- (a) A. M. Santana, A. E. Mauro, H. E. Zorel Jr., M. P. D. Mattioli and V. A. De Lucca Neto, J. Therm. Anal. Cal., 2002, 67, 425–431 Search PubMed; (b) S. R. Ananias, A. E. Mauro, K. Zutin, C. M. C. Picchi and R. H. A. Santos, Transition Met. Chem., 2004, 29, 284–290 CrossRef CAS.
- I. Dance and M. Scudder, J. Chem. Soc., Dalton Trans., 2000, 1579–1585 RSC.
- N. M. Chaignon, I. J. S. Fairlamb, A. R. Kapdi, R. J. K. Taylor and A. C. Whitwood, J. Mol. Cat. A: Chem., 2004, 219, 191–199 CrossRef CAS.
- R. R. Tykwinski, Angew. Chem. Int. Ed., 2003, 42, 1566–1568 CrossRef CAS.
- (a) P. Nguyen, Z. Yuan, L. Agocs, G. Lesley and T. B. Marder, Inorg. Chim. Acta, 1994, 220, 289–296 CrossRef CAS; (b) E. V. Tretyakov, D. W. Knight and S. F. Vasilevsky, J. Chem. Soc., Perkin Trans. 1, 1999, 24, 3713–3720 RSC.
- (a) N. E. Leadbeater and B. J. Tominack, Tetrahedron Lett., 2003, 44, 8653–8656 CrossRef CAS; (b) D. Mery, K. Heuze and D. Astruc, Chem. Commun., 2003, 1934–1935 RSC; (c) A. Soheili, J. Albaneze-Walker, J. A. Murry, P. G. Dormer and D. L. Hughes, Org. Lett., 2003, 5, 4191–4194 CrossRef CAS; (d) Y. D. Ma, C. Song, W. Jiang, Q. S. Wu, Y. Wang, X. Y. Liu and M. B. Andrus, Org. Lett., 2003, 5, 3317–3319 CrossRef CAS; (e) D. Gelman and S. L. Buchwald, Angew. Chem. Int. Ed., 2003, 42, 5993–5996 CrossRef CAS.
- M. T. Reetz and J. G. de Vries, Chem. Commun., 2004, 1559–1563 RSC.
- A. H. M. de Vries, J. M. C. A. Mulders, J. H. M. Mommers, H. J. W. Henderickx and J. G. de Vries, Org. Lett., 2003, 5, 3285–3288 CrossRef CAS.
- During the preparation of this manuscript a very useful and notable protocol has been developed for phosphine removal using Cu(I) salts, see: B. H. Lipshutz, B. Frieman and H. Birkedal, Org. Lett., 2004, 6, 2305–2308 Search PubMed.
- S. M. Nobre, S. I. Wolke, R. G. da Rosa and A. L. Monteiro, Tetrahedron Lett., 2004, 45, 6527–6530 CrossRef CAS.
- PEO 3350 (Mw = 10,000) was used in the procedure reported by Monteiro and co-workers (ref. 31). In our studies, we employed PEO with a Mw = 100,000.
- I. Aiello, A. Crispini, M. Ghedini, M. La Deda and F. Barigelletti, Inorg. Chim. Acta, 2000, 308, 121–128 CrossRef CAS.
- SAINT, Bruker AXS Inc, Madison, WI, V. 6.22, 2003 Search PubMed.
- G. M. Sheldrick, SADABS, Program for area detector absorption correction, University of Göttingen, Germany, 1966 Search PubMed.
- G. M. Sheldrick, SHELX-97, Programs for Crystal Structure Analysis, University of Göttingen, Germany, 1998, release 97-2 Search PubMed.
- E. H. Niemelä, A. F. Lee and I. J. S. Fairlamb, Tetrahedron Lett., 2004, 45, 461–465 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2004 |